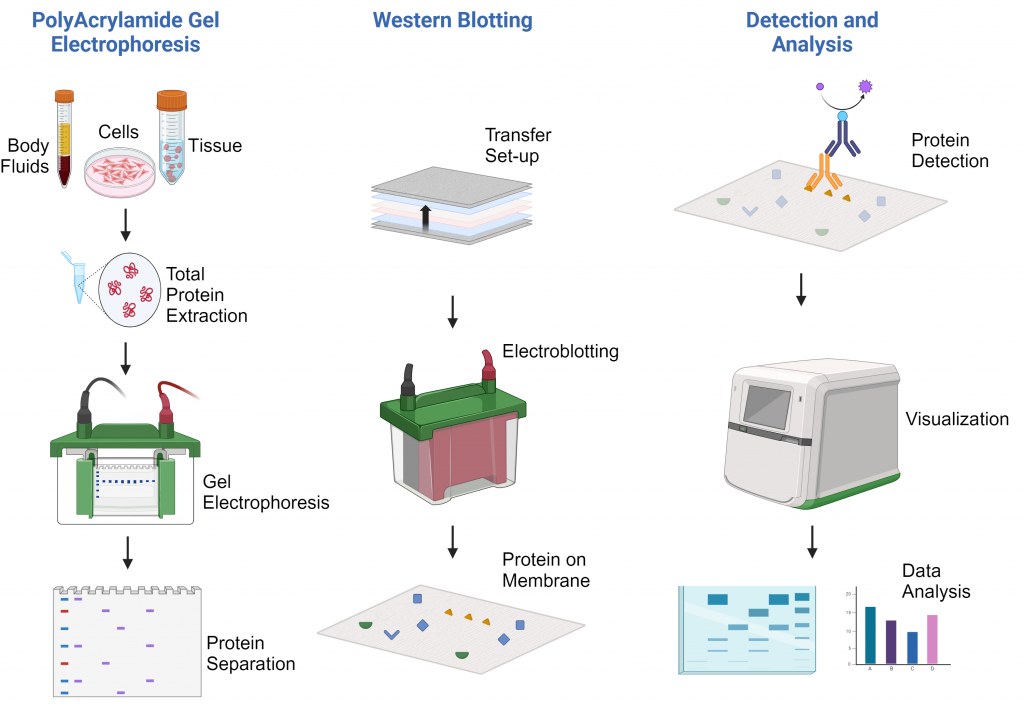
Protocolo de Western Blot

Principio de la Técnica
El principio de la técnica de Western blotting involucra varios pasos críticos:
-
Separación de proteínas: Las proteínas se separan por tamaño mediante SDS-PAGE.
-
Transferencia: Las proteínas separadas se transfieren a una membrana sólida (nitrocelulosa o PVDF).
-
Bloqueo: Las superficies de la membrana se bloquean para evitar la unión no específica.
-
Detección: Anticuerpos primarios específicos reconocen las proteínas objetivo, seguidos de anticuerpos secundarios marcados.
-
Visualización: Los complejos proteína-anticuerpo se visualizan mediante reacciones quimioluminiscentes, fluorescentes o colorimétricas.
El éxito de la técnica depende en gran medida de la especificidad de los anticuerpos y de la integridad de la preparación de las muestras.
El primer paso importante que no debe pasarse por alto al realizar un Western blot es la preparación de la muestra. Para obtener resultados reproducibles, debe llevarse a cabo una extracción y purificación eficiente de proteínas utilizando un método de homogenización adecuado que pueda liberar de manera eficiente el contenido intracelular de la célula mediante la ruptura de las membranas celulares.
Aislamiento de Muestras de Proteínas a Partir de Diferentes Tipos de Muestras
La extracción y el aislamiento de proteínas son pasos críticos en una amplia gama de técnicas bioquímicas y de biología molecular, incluidos Western blotting, espectrometría de masas, ensayos enzimáticos e inmunoprecipitación. El éxito de las aplicaciones posteriores depende en gran medida de la calidad, integridad y representatividad de las muestras de proteínas aisladas.
Las muestras biológicas como células cultivadas, tejidos, bacterias, levadura, y material vegetal presentan desafíos únicos para la extracción de proteínas. Por lo tanto, elegir una estrategia de aislamiento apropiada es esencial para maximizar el rendimiento, preservar la funcionalidad de las proteínas y minimizar la degradación o modificación.
1. Homogenización
-
Elija un método de homogenización según el tipo de muestra:
-
Ultrasonido (para muestras delicadas)
-
Prensa francesa (para bacterias, levaduras)
-
Molienda con perlas de vidrio (para células duras, hongos)
-
Homogeneizador Dounce/Potter-Elvehjem (para tejidos blandos)
-
Molienda manual con mortero/pistilo (para tejidos congelados)
-
-
Objetivo: Lisis celular eficiente sin degradación de proteínas.
2. Manipulación de Muestras Posterior al Tratamiento
-
Después de la exposición a xenobióticos o manipulación genética (por ejemplo, silenciamiento de siRNA):
-
Congelar inmediatamente las muestras en nitrógeno líquido
-
Lisado inmediatamente para prevenir la actividad de proteasas endógenas.
-
-
Siempre mantener las muestras frías durante el procesamiento.
3. Gestión del Ciclo de Congelación/Descongelación
-
Evitar ciclos múltiples de congelación/descongelación para proteger la integridad de las proteínas.
-
Alícuote las muestras para evitar descongelaciones innecesarias.
-
4. Selección de Buffer de Lisis
a. Alinear el buffer con la ubicación de la proteína objetivo:
-
Proteínas citosólicas → Buffer de lisis suave (por ejemplo, NP-40, Triton X-100)
-
Proteínas unidas a membranas → Buffers más fuertes (por ejemplo, RIPA)
-
Proteínas nucleares/mitocondriales → Buffers especializados (por ejemplo, Buffer hipotónico, Buffer de Homogenización de Drosophila)
b. Alinear el buffer con la sensibilidad del anticuerpo:
-
Detección de proteínas nativas → Condiciones suaves/detergente no iónico o sin detergente
-
Detección de proteínas desnaturalizadas → Condiciones fuertes/desnaturalizantes
-
Incluir inhibidores de proteasas y fosfatasas en todos los buffers de lisis.
c. Algunos ingredientes comunes y por qué se utilizan:
-
HEPES: Mantiene la estabilidad del pH.
-
Sacarosa /Mannitol: Estabilizadores osmóticos para prevenir la ruptura de orgánulos.
-
EDTA /EGTA: Quelan cationes divalentes para inhibir metaloproteasas y prevenir la degradación.
-
Inhibidores de proteasas y fosfatasas: Protegen las proteínas de la degradación enzimática.
5. Notas críticas sobre la fraccionamiento de muestras
a. Cuidado con la pérdida de proteínas durante la centrifugación:
-
Verifique si la proteína objetivo se retiene en el pellet (desperdicios).
-
Ejemplo: Hasta el 50% de la miosina y ~66% de CSQ2 se perdieron en los desperdicios si no se verificó.
b. Siempre verifique si la proteína de interés es:
-
Soluble (citosólica)
-
Insoluble (citosquelética)
-
Unida a membranas (orgánulos)
Separación electroforética
Tras la extracción y cuantificación de proteínas, la siguiente fase crítica en el análisis proteómico es la separación electroforética. La electroforesis en gel sigue siendo una técnica fundamental para mejorar la selectividad y sensibilidad en la investigación del proteoma, permitiendo la resolución de mezclas complejas de proteínas en función de sus propiedades fisicoquímicas.

Se han desarrollado varias matrices de gel, incluyendo agarosa, almidón y poliacrilamida, para aplicaciones electroforéticas. Entre ellas, la poliacrilamida es la matriz de elección para el análisis de proteínas debido a su estabilidad mecánica, inercia química y la capacidad de regular con precisión la polimerización con bisacrilamida. Una proporción comúnmente utilizada de acrilamida:bisacrilamida de 37.5:1 permite la generación de geles robustos con tamaños de poro reproducibles, adecuados para la fraccionamiento de proteínas de alta resolución.
Existen dos formas principales de electroforesis en gel de poliacrilamida (PAGE) ampliamente utilizadas: PAGE nativa y SDS-PAGE desnaturalizante.
-
PAGE nativa preserva las estructuras terciarias y cuaternarias de las proteínas, permitiendo su separación en función de una combinación de carga neta, tamaño hidrodinámico y conformación. En sistemas de tampones alcalinos, la mayoría de las proteínas adquieren una carga negativa neta, permitiendo su migración hacia el ánodo sin desnaturalización. Una ventaja importante de PAGE nativa es la retención de la actividad enzimática e interacciones nativas entre proteínas durante la separación.
-
SDS-PAGE (electroforesis en gel de poliacrilamida con dodecil sulfato sódico) es el método más comúnmente utilizado para separar proteínas desnaturalizadas basándose únicamente en el peso molecular. El detergente aniónico SDS se une uniformemente a lo largo de la columna vertebral de los polipéptidos, impartiendo una carga negativa consistente proporcional a la longitud de la proteína. Bajo condiciones desnaturalizantes (generalmente con calentamiento en 0.1% SDS), las proteínas se linealizan y adoptan una forma extendida tipo barra, lo que permite la migración dependiente del tamaño a través de la matriz de poliacrilamida. Las proteínas más pequeñas atraviesan el gel más rápidamente que las de mayor tamaño. Además, SDS-PAGE se beneficia de los principios de isotachoforesis, concentrando las muestras de proteínas en bandas definidas y apretadas, mejorando así la resolución (≥ concentración diez veces observada en configuraciones de laboratorio controladas).
La atención al proceso de preparación de la muestra, composición de los tampones y características del gel es esencial para lograr una separación óptima y reproducibilidad. Los componentes de los tampones típicos de muestra y sus respectivas funciones se resumen en la Tabla 1.
Tabla 1. Buffers comúnmente utilizados para la preparación de muestras. Ubicación de la proteína Buffer Propósito de los componentes (concentraciones típicas indicadas entre paréntesis)
| Ubicación de la proteína | Buffer | Propósito de los componentes (concentraciones típicas entre paréntesis) |
| Célula completa | Buffer no desnaturalizante y no iónico: Buffer NP-40: 50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40 o Triton X-100 |
Tris-HCl (10–50 mM): tamponamiento de la solución (ayuda a mantener la estabilidad de las proteínas) NaCl (50–150 mM): reduce la unión no específica, mantiene la fuerza iónica NP-40/Triton X-100 (0.1–1%): detergentes no iónicos, solubiliza proteínas citosólicas y de membrana sin desnaturalizarlas, permeabiliza membranas e incrementa la solubilidad de las proteínas previniendo la agregación |
| Buffer desnaturalizante: Buffer RIPA: 150 mM NaCl, 1% NP-40 o Triton X-100, 0.5% desoxicolato sódico, 0.1% SDS, 50 mM Tris, pH 8.0 |
Desoxicolato sódico (0.1–0.5%): detergente iónico, lisis de células y solubilización de componentes celulares y de membrana SDS (0.1–1%): detergente iónico, causa disrupción de membranas y linealiza las proteínas uniéndose a ellas |
|
| Citosplásmica (soluble) |
20 mM Tris-HCl, pH 7.5 o buffer NP-40 | Ver arriba |
| Citosplásmica (unida al citoesqueleto) |
Buffer PIPES-Triton: 10 mM PIPES, 50 mM KCl, 1% Triton X-100, 10 mM EGTA, 3 mM MgCl2, 2 M glicerol |
PIPES (10 M): tamponamiento de la solución KCl (50–150 mM): reduce la unión no específica, mantiene la fuerza iónica EGTA (1–3 mM): inhibición de metaloproteasas dependientes de Ca2+ MgCl2 (1–5 mM): reduce la unión no específica, junto con KCl tiene un efecto estabilizador sobre algunos complejos Glicerol (1–10%): estabiliza las proteínas |
| Unida a membranas | Buffer RIPA | Ver arriba |
| Mitocondrias | Buffer RIPA | Ver arriba |
| Nuclear | Buffer RIPA | Ver arriba; nota: NP-40 y Triton X-100 no pueden lisar membranas nucleares |
| Ubicación de la proteína | Buffer |
El porcentaje de gel requerido depende del tamaño de la proteína de interés:
| Tamaño de la proteína | Porcentaje de gel |
| 4–40 kDa | 20% |
| 12–45 kDa | 15% |
| 10–70 kDa | 12% |
| 15–100 kDa | 10% |
| 25–100 kDa | 8% |
| 60-210 kDa | 5% |
También se pueden utilizar geles de gradiente.
| Tamaño de la proteína | Porcentaje de gel |
| 5-200 kDa | Gradiente 4-12 |
| 4-200 kDa | Gradiente 4-20 |
| 3.5-110 kDa | Gradiente 10-20 |
Sistemas de tampones continuos versus discontinuos
Los sistemas de separación electroforética pueden clasificarse ampliamente en sistemas de tampones continuos y discontinuos (también llamados sistemas multipásicos o isotachoforesis), cada uno caracterizado por configuraciones de tampones y dinámicas de separación distintas.
1. Sistemas de tampones continuos:
-
Same buffer ions and pH across sample, gel, and reservoirs.
-
Gel made of a single acrylamide concentration.
-
Proteins separate directly in the gel without stacking.
-
Punto clave: La resolución depende en gran medida de cargas de muestra pequeñas y concentradas.
2. Sistemas de tampones discontinuos (Multipásicos):
-
Dos tampones diferentes con pH y fuerza iónica distintas:
-
Gel de apilamiento: pH ~6.8, bajo Tris–HCl (0.125 M), poros grandes.
-
Gel de resolución: pH ~8.8, alto Tris–HCl (0.375 M), poros pequeños.
-
-
Buffer de corrida (por ejemplo, Tris-glicina) introducido por encima del gel de apilamiento.
3. Mecanismo de separación:
-
Los iones Cl⁻ actúan como iones principales; el glicinato como iones secundarios.
-
Las proteínas se concentran en bandas compactas en el gel de apilamiento ("efecto de apilamiento").
-
En el gel de resolución, las proteínas se separan por peso molecular a medida que se convierten en los iones principales.
4. Ventajas de los Sistemas Discontinuos:
-
Permite cargar volúmenes mayores de muestras diluidas.
-
Mejor resolución debido al apilamiento y las diferentes tamaños de poro en los geles.
-
Separación más efectiva basada en el peso molecular.
Transferencia de Proteínas
Después de la electroforesis, las proteínas se transfieren de un gel de poliacrilamida a una membrana. Este paso es crucial para inmovilizar las proteínas para su posterior detección y análisis (por ejemplo, western blotting).
1. Membranas Utilizadas para la Transferencia de Proteínas
Existen diferentes membranas disponibles, cada una con ventajas específicas:
-
Nitrocelulosa es la membrana más comúnmente utilizada. Tiene una alta afinidad por la unión de proteínas, puede inmovilizar tanto proteínas como glucoproteínas, y es compatible con métodos de detección quimioluminiscente, cromogénica y fluorescente. Sin embargo, es quebradiza debido a que se deriva de la celulosa tratada con ácido nítrico.
-
PVDF (Poli(fluoruro de vinilideno)) ofrece una mejor resistencia mecánica que la nitrocelulosa, lo que la hace ideal para despojar y reprobar o para secuenciar proteínas. Sin embargo, el PVDF a veces puede generar un mayor fondo de tinción, lo que requiere optimización adicional.
2. Métodos para la Transferencia de Proteínas
Las proteínas se pueden transferir del gel a la membrana utilizando varios métodos:
-
Transferencia capilar se basa en la acción capilar pasiva.
-
Transferencia por difusión permite que las proteínas se difundan lentamente hacia la membrana.
-
Electrotransferencia es el método más rápido y completo para la transferencia.
Entre estos, la electrotransferencia es el método preferido por su velocidad y eficiencia.
Tipos de Electrotransferencia
Existen tres tipos de electrotransferencia basados en el método de uso de los tampones:
-
Electrotransferencia seca utiliza componentes secos sin líquido añadido.
-
Electrotransferencia semi-seca utiliza papeles filtrantes empapados con tampón de transferencia entre la membrana y el gel.
-
Electrotransferencia húmeda sumerge todo el sándwich gel-membrana en un tampón de transferencia, lo que es especialmente recomendable para la transferencia de proteínas grandes.
1. Principios de la Electrotransferencia
Durante la electrotransferencia:
-
Se aplica un campo eléctrico perpendicular al gel.
-
Las proteínas, al estar cargadas, migran fuera del gel hacia la membrana.
-
El gel debe estar frente al electrodo negativo (cátodo), mientras que la membrana debe estar frente al electrodo positivo (ánodo).
-
Para asegurar una transferencia adecuada, el gel y la membrana se deben colocar firmemente juntos, con papeles filtrantes y almohadillas de fibra a ambos lados para evitar la formación de burbujas, lo que podría causar una transferencia desigual.
a. Tampones de Transferencia
Los tampones de transferencia ayudan a mantener la estructura de las proteínas y mejoran su movimiento durante la transferencia. Los tampones comunes incluyen:
-
Tampón de Towbin (25 mM Tris, 192 mM glicina, 20% metanol, pH 8.3) se utiliza ampliamente para transferencia húmeda.
-
Tampón de Bjerrum Schafer-Nielsen (48 mM Tris, 39 mM glicina, pH 9.2, 20% metanol) es preferido para la transferencia semi-seca.
-
Tampón CAPS (60 mM Tris y 40 mM CAPS) se usa especialmente para proteínas con puntos isoeléctricos altos.
El metanol en el tampón ayuda a prevenir la hinchazón del gel y mejora la unión de las proteínas a la membrana. Sin embargo, en algunos casos, los tampones sin metanol aún pueden producir señales de detección fuertes.
El papel de los componentes del tampón de transferencia se describe en la Tabla 2.
Tabla 2. Composición de los tampones de corrida y transferencia y el papel de cada componente
| Componente potencial | Función |
| Tampones de corrida | |
| Acetato | Buena separación en un rango de masa más alto (100–500 kDa) |
| Glicina | Proporciona iones glicinato para formar el extremo de arrastre durante la electroforesis en el gel de apilamiento |
| MES | Necesario para mantener un pH relativamente constante. Proporciona iones MES para formar el extremo de arrastre durante la electroforesis. El MES resulta en una separación más grande en un rango de masa más bajo (<50 kDa). El tampón MES SDS es más rápido que el tampón MOPS SDS debido al pKa más bajo del MES en comparación con el MOPS |
| MOPS | Necesario para mantener un pH relativamente constante. Proporciona iones MOPS para formar el extremo de arrastre durante la electroforesis. El MOPS da una mejor separación en rangos de masa medios a altos. La diferencia en el movimiento iónico de MES y MOPS impacta el apilamiento, lo que resulta en una diferencia en el rango de separación de proteínas entre los tampones MES y MOPS |
| SDS | Ayuda a mantener las proteínas con una carga neta negativa |
| Tricina | Necesario para mantener un pH relativamente constante. La tricina sustituye a la glicina en el tampón de corrida, proporcionando iones tricina para formar el extremo de arrastre durante la electroforesis. La tricina resulta en un apilamiento y resolución más eficientes de proteínas de bajo peso molecular. Buena para separar proteínas de bajo peso molecular (0.5–50 kDa) |
| Tris-HCl | Necesario para mantener un pH relativamente constante. Proporciona iones de cloruro que forman el extremo principal durante la electroforesis en el gel de apilamiento. Los iones de hidrógeno conducen electricidad |
| Tampones de transferencia | |
| Tris-HCl | Necesario para mantener un pH relativamente constante |
| Glicina | En ausencia de metanol, ayuda a prevenir la hinchazón del gel |
| Metanol | Previene la hinchazón del gel durante la transferencia y mejora la unión de las proteínas a la nitrocelulosa. Elimina el SDS de las proteínas permitiendo una mejor unión a las membranas de nitrocelulosa. Algunos laboratorios ya no incluyen metanol en los tampones de transferencia, ya que los resultados sin metanol no fueron significativamente diferentes de los obtenidos con metanol. Algunos protocolos utilizan 10% de etanol en lugar de 20% de metanol |
| SDS | El SDS (hasta 0.1%) en el tampón de transferencia aumenta la eficiencia de la transferencia de proteínas, especialmente para proteínas grandes, pero puede reducir la cantidad de proteína que se une a la membrana. En membranas de nitrocelulosa con tamaños de poro más grandes (0.45 µm), las proteínas pequeñas desnaturalizadas por SDS pueden atravesar la membrana |
| CAPS | Necesario para mantener un pH relativamente constante. Recomendado para la transferencia de proteínas de alto peso molecular (>150 kDa) |
| CAPS: (Ácido (Ciclohexilamino)-L-propanosulfónico); MES: Ácido 2-(N-morfolino) etanosulfónico; MOPS: Ácido 3-(N-morfolino) propanosulfónico; SDS: Lauril sulfato sódico; Tris-HCl: Tris cloruro de hidrógeno. |
|
b. Consideraciones Importantes
-
Las proteínas con un peso molecular muy bajo (<10 kDa) pueden transferirse de manera ineficiente.
-
Si el pH del tampón de transferencia es inferior al punto isoeléctrico de la proteína, las proteínas pueden migrar hacia atrás.
-
Los tampones especializados (como los tampones a base de CAPS) suelen ser necesarios para transferir eficientemente proteínas con un alto punto isoeléctrico.
Paso de Bloqueo en Western Blotting
Después de una transferencia exitosa de proteínas a la membrana, el bloqueo es un paso crítico para asegurar la especificidad y claridad de la inmunodetección posterior.

1. Propósito e Importancia
-
El bloqueo previene la unión inespecífica de los anticuerpos a la membrana, minimizando así el ruido de fondo y eliminando señales falsas positivas.
-
Mejora la precisión y confiabilidad de la detección de proteínas durante el western blotting.
2. Procedimiento de Bloqueo
-
La membrana se incuba durante 1 hora a temperatura ambiente en una solución de bloqueo que contiene:
-
Albúmina sérica bovina (BSA) o leche en polvo descremada, diluida en:
-
Solución salina tamponada con Tris con 0.1% Tween 20 (TBST), o
-
Solución salina tamponada con fosfato con 0.1% Tween 20 (PBST).
-
-
-
Las proteínas presentes en BSA y la leche (principalmente caseína y proteínas del suero) se unen a los sitios no ocupados de la membrana, evitando así interacciones inespecíficas entre la membrana y los anticuerpos primarios o los anticuerpos secundarios.
3. Características de la Membrana y el Papel del Tween 20
-
Membranas de nitrocelulosa y PVDF exhiben altas afinidades de unión de proteínas principalmente a través de interacciones hidrofóbicas.
-
Si bien las proteínas de BSA y la leche pueden asociarse débilmente con las proteínas transferidas, la adición de Tween 20 en el tampón de bloqueo reduce estas interacciones inespecíficas, asegurando un bloqueo más efectivo.
Incubación de Anticuerpos y Detección en Western Blotting
1. El Papel de la Incubación de Anticuerpos en Western Blotting
Después del bloqueo de la membrana para prevenir la unión inespecífica, western blotting avanza al paso crucial de la incubación de anticuerpos. Primero, la membrana se incuba con un anticuerpo primario que se une específicamente a la proteína objetivo. Después de lavar los anticuerpos no unidos con tampones como TBST o PBST, se introduce un anticuerpo secundario. Este anticuerpo secundario reconoce al anticuerpo primario y normalmente está conjugado a una enzima como HRP (peroxidasa de rábano picante) o Fosfatasa alcalina (AP), lo que permite la posterior detección de la señal.
-
Anticuerpos Primarios: Esenciales para la unión específica y selectiva a las proteínas objetivo.
-
Anticuerpos Secundarios: Permiten la detección y amplificación de la señal.
Consejo: Es importante optimizar la concentración de anticuerpos para maximizar la sensibilidad y reducir el fondo. Nuestro soporte técnico puede ayudarte con la optimización del protocolo.
Los pasos de lavado son críticos para eliminar los anticuerpos no unidos. Sin embargo, un lavado excesivo (>20 minutos) puede debilitar la señal, ¡por lo que es clave el tiempo equilibrado!
2. Métodos de Detección de Señales
Una vez que la proteína objetivo se une a los anticuerpos marcados, se debe detectar la señal. Existen varios métodos disponibles:
a. Quimioluminiscencia (ECL)
Quimioluminiscencia mejorada (ECL) es el método de detección más popular. Utilizando anticuerpos secundarios conjugados con HRP y sustratos ECL, se genera luz que se captura ya sea en película o mediante un imagen digital.
-
Alta sensibilidad
-
Amplio rango dinámico
-
Ideal para detectar proteínas de baja abundancia
Productos recomendados:
Anticuerpos Secundarios Conjugados con HRP
Kits de Sustrato ECL
Kits de Detección ECL
b. Detección por Fluorescencia
Los anticuerpos secundarios conjugados con colorantes fluorescentes permiten la detección multiplex — lo que significa que se pueden visualizar varias proteínas a la vez usando diferentes fluoróforos.
-
Adecuado para detección simultánea de múltiples objetivos
-
Excelente rango dinámico y cuantificación
-
Mejor al usar fluoróforos en el rango infrarrojo
Productos recomendados:
Anticuerpos Secundarios (Visible e Infrarrojo)
Sistemas de Imagen Fluorescente
Anticuerpos Fluorescentes
c. Detección Cromogénica
Los métodos cromogénicos utilizan reacciones enzimáticas para generar un precipitado coloreado en la membrana, visible a simple vista.
- Adecuado cuando el equipo de imagen es limitado.
-
Visualización sencilla
-
No requiere equipos especializados
-
Ideal para laboratorios educativos y de investigación básica
Productos recomendados:
Kits de Sustrato Cromogénico (por ejemplo, BCIP/NBT)
d. Detección Radiactiva (Menos Común Hoy en Día)
Los sondas radiactivas fueron ampliamente utilizadas en western blotting pero ahora han sido reemplazadas por métodos más seguros y igualmente sensibles como ECL y fluorescencia. La selección del método de detección adecuado es crucial para el éxito de tu Western blot y depende de la sensibilidad, especificidad y tipo de análisis que requieras:
-
¿Máxima sensibilidad? → Elige detección por quimioluminiscencia mejorada (ECL)
ECL ofrece alta sensibilidad y gran rango dinámico, siendo ideal para detectar proteínas de baja abundancia. La intensidad de la señal se puede capturar en películas de rayos X o sistemas de imágenes digitales. -
¿Análisis multiplex (detección de múltiples objetivos simultáneamente)? → Elige detección por fluorescencia
Los anticuerpos secundarios fluorescentes permiten la detección simultánea de múltiples proteínas sin necesidad de retirar y reprobar las membranas. Las señales son altamente estables, cuantitativas e ideales para estudios comparativos. -
¿Visualización simple y rápida? → Elige detección cromogénica
Los sistemas cromogénicos utilizan reacciones enzima-sustrato para producir un precipitado coloreado directamente en la membrana. Aunque menos sensibles que ECL o fluorescencia, son fáciles de usar y no requieren equipo especializado.
Además, la elección de anticuerpos primarios y secundarios, así como de sustratos o reactivos de detección, debe ser compatible con el método de detección seleccionado. Emparejar correctamente es esencial para lograr señales específicas y fuertes mientras minimizas el ruido de fondo.