Protocolo Western Blot

Princípio da técnica
O princípio do Western blotting envolve várias etapas críticas:
-
Separação de proteínas: As proteínas são separadas por tamanho através de SDS-PAGE.
-
Transferência: As proteínas separadas são transferidas para uma membrana sólida (nitrocelulose ou PVDF).
-
Bloqueio: As superfícies da membrana são bloqueadas para impedir a ligação não específica.
-
Detecção: Anticorpos primários específicos reconhecem as proteínas alvo, seguidos por anticorpos secundários marcados.
-
Visualização: Os complexos proteína-anticorpo são visualizados através de reações quimioluminescentes, fluorescentes ou colorimétricas.
O sucesso da técnica depende muito da especificidade do anticorpo e da integridade da preparação da amostra.
O primeiro passo importante que não deve ser esquecido ao realizar a transferência Western é a preparação da amostra. Para obter resultados reproduzíveis, deve-se realizar uma extração e purificação eficiente das proteínas utilizando um método de homogeneização adequado que possa liberar eficientemente o conteúdo intracelular da célula através da ruptura das membranas celulares.
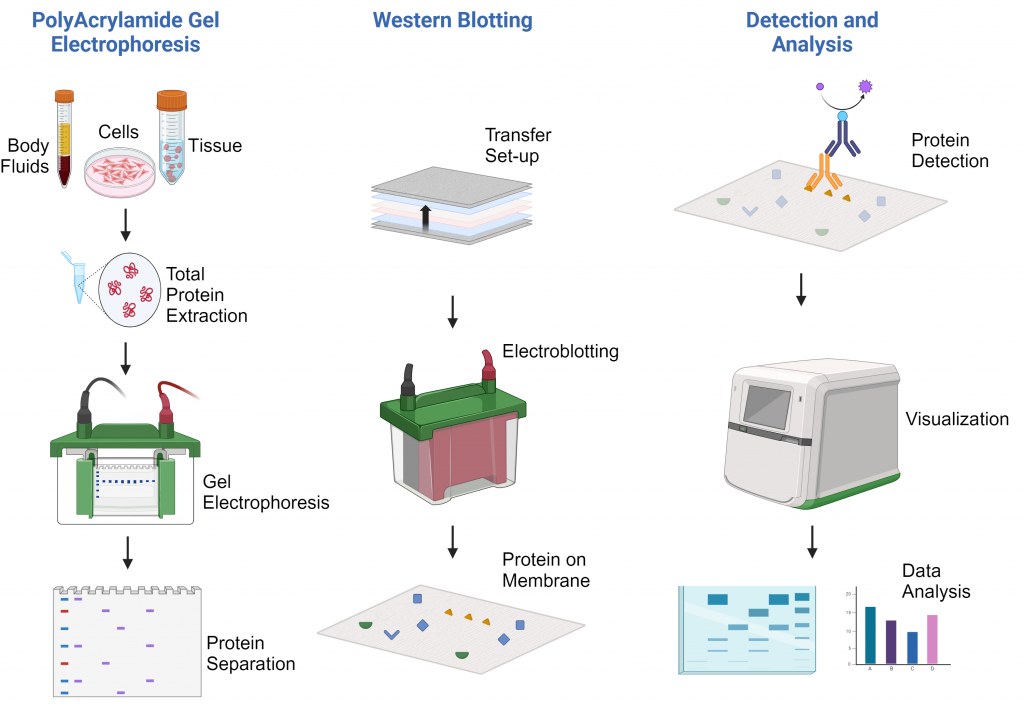
Isolamento de proteínas de diferentes tipos de amostras
A extração e o isolamento de proteínas são etapas iniciais essenciais em uma ampla gama de técnicas bioquímicas e de biologia molecular, incluindo Western blotting, espectrometria de massa, ensaios enzimáticos e imunoprecipitação. O sucesso das aplicações a jusante depende muito da qualidade, integridade e representatividade das amostras de proteínas isoladas.
Amostras biológicas, tais como células cultivadas, tecidos, bactérias, leveduras e material vegetal, apresentam desafios únicos para a extração de proteínas. Portanto, é essencial escolher uma estratégia de isolamento adequada para maximizar o rendimento, preservar a funcionalidade das proteínas e minimizar a degradação ou modificação.
1. Homogeneização
-
Escolha um método de homogeneização com base no tipo de amostra:
- Ultrassonificação (para amostras delicadas)
- Prensa francesa (para bactérias, leveduras)
- Moagem com esferas de vidro (para células resistentes, fungos)
- Homogeneizador Dounce/Potter-Elvehjem (para tecidos moles)
- Moagem manual com almofariz/pilão (para tecidos congelados)
-
Objetivo: Lise celular eficiente sem degradação de proteínas.
2. Manuseamento de amostras pós-tratamento
-
Após exposição a xenobióticos ou manipulação genética (por exemplo, silenciamento por siRNA):
-
Congele imediatamente as amostras em nitrogénio líquido.
-
Lisar imediatamente para evitar a atividade da protease endógena.
-
- Mantenha sempre as amostras refrigeradas durante o processamento.
3. Gestão do ciclo de congelamento/descongelamento
-
Evite vários ciclos de congelamento/descongelamento para proteger a integridade das proteínas..
-
Divida as amostras em alíquotas para evitar descongelamento desnecessário.
-
4. Seleção do tampão de lise
a.Correspondência do buffer com a localização da proteína alvo:
-
Proteínas citosólicas → Tampão de lise suave (por exemplo, NP-40, Triton X-100)
-
Proteínas ligadas à membrana → Tampões mais fortes (por exemplo, RIPA)
-
Proteínas nucleares/mitocondriais → Tampões especializados (por exemplo,Tampão hipotónico, Tampão de homogeneização de Drosophila)
b.Combinar o tampão com a sensibilidade do anticorpo:
-
Detecção de proteínas nativas → Detergente suave/ não iônico ou sem detergente
-
Detecção de proteínas desnaturadas → Condições fortes/desnaturantes
-
Inclua inibidores de protease e fosfatase em todos os tampões de lise.
c.Alguns ingredientes comuns e por que são usados:
-
HEPES: Mantém a estabilidade do pH.
-
Sacarose / Manitol: Estabilizadores osmóticos para prevenir a ruptura das organelas.
-
EDTA / EGTA: Chelates cátions divalentes para inibir metaloproteases e prevenir a degradação.
-
Protease e fosfatase inibidores: Protegem as proteínas da degradação enzimática.
5. Notas críticas sobre o fraccionamento de amostras
a.Cuidado com a perda de proteínas durante a centrifugação:
-
Teste se a proteína alvo está retida no sedimento (detritos).
-
Exemplo: Até 50% da miosina e ~66% do CSQ2 foram perdidos nos detritos se não fossem verificados.
b.Verifique sempre se a proteína de interesse é:
-
Solúvel (citoplasmático)
-
Insolúvel (citoesquelético)
-
Ligado à membrana (organelar)
Separação eletroforética
Após a extração e quantificação das proteínas, a próxima fase crítica na análise proteómica é a separação eletroforética. A eletroforese em gel continua a ser uma técnica fundamental para aumentar a seletividade e a sensibilidade na investigação proteómica, permitindo a resolução de misturas complexas de proteínas com base nas suas propriedades físico-químicas.

Várias matrizes de gel — incluindo agarose, amido e poliacrilamida — foram desenvolvidas para aplicações eletroforéticas. Entre elas, a poliacrilamida é a matriz preferida para análise de proteínas devido à sua estabilidade mecânica, inércia química e capacidade de regular com precisão a polimerização com bisacrilamida. Uma proporção comum de acrilamida:bisacrilamida de 37,5:1 permite a geração de géis robustos com tamanhos de poros reproduzíveis, adequados para fraccionamento de proteínas de alta resolução.
Duas formas principais de eletroforese em gel de poliacrilamida (PAGE) são amplamente utilizadas: PAGE nativa e SDS-PAGE desnaturante.
-
A PAGE nativa preserva as estruturas terciárias e quaternárias das proteínas, permitindo a sua separação com base numa combinação de carga líquida, tamanho hidrodinâmico e conformação. Em sistemas tampão alcalinos, a maioria das proteínas adquire uma carga líquida negativa, permitindo a migração para o ânodo sem desnaturação. Uma grande vantagem da PAGE nativa é a retenção da atividade enzimática e das interações proteína-proteína nativas durante a separação..
-
SDS-PAGE (eletroforese em gel de dodecil sulfato de sódio-poliacrilamida) é o método mais comumente empregado para separar proteínas desnaturadas com base exclusivamente no peso molecular. O detergente aniônico SDS liga-se uniformemente ao longo da estrutura do polipeptídeo, conferindo uma carga negativa consistente proporcional ao comprimento da proteína. Em condições de desnaturação (normalmente com aquecimento em SDS a 0,1%), as proteínas são linearizadas e adotam uma forma alongada semelhante a uma haste, permitindo a migração dependente do tamanho através da matriz de poliacrilamida. As proteínas mais pequenas atravessam o gel mais rapidamente do que as suas contrapartes maiores. Além disso, a SDS-PAGE beneficia dos princípios da isotacóforese, concentrando as amostras de proteínas em bandas estreitas e definidas, melhorando assim a resolução (concentração ≥ dez vezes superior observada em condições laboratoriais controladas).
É essencial prestar atenção à preparação da amostra, à composição do tampão e às características do gel para obter uma separação e reprodutibilidade ideais. Os componentes dos tampões de amostra típicos e as suas respetivas funções estão resumidos na Tabela 1.
Tabela 1. Tampões de preparação de amostras comumente usados. Localização da proteína Tampão Finalidade
| Localização da proteína | Amortecedor | Finalidade dos componentes (concentrações típicas indicadas entre parênteses) |
| Célula inteira |
Tampão não desnaturante, não iónico: tampão NP-40: 50 mM Tris pH 8,0, 150 mM NaCl, 1% NP-40 ou Triton X-100 |
Tris-HCl (10–50 mM): tamponamento da solução (ajuda a manter a estabilidade das proteínas) NaCl (50–150 mM): reduz a ligação não específica, mantém a força iônica NP-40/Triton X-100 (0,1–1%): detergentes não iônicos, solubilizam proteínas citosólicas e de membrana sem denaturá-las, permeabilizam membranas e aumentam a solubilidade das proteínas, impedindo a agregação |
|
Tampão desnaturante: Tampão RIPA: 150 mM NaCl, 1% NP-40 ou Triton X-100, 0,5% de desoxicolato de sódio, 0,1% SDS, 50 mM Tris, pH 8,0 |
Desoxicolato de sódio (0,1–0,5%): detergente iônico, lisa as células e solubiliza componentes celulares e membranares SDS (0,1–1%): detergente iônico, causa ruptura da membrana e lineariza proteínas ao ligar-se a elas |
|
| Citoplasmático (solúvel) |
20 mM Tris-HCl, pH 7,5 ou tampão NP-40 | Veja acima |
| Citoplasmático (ligado ao citoesqueleto) |
PIPES-Triton buffer: 10 mM PIPES, 50 mM KCl, 1% Triton X-100, 10 mM EGTA, 3 mM MgCl2, 2 M glicerol |
TUBOS (10 M): tamponamento da solução KCl (50–150 mM): reduz a ligação não específica, mantém a força iônica EGTA (1–3 mM): inibição das metaloproteases Ca2+ MgCl2 (1–5 mM): reduz a ligação não específica, juntamente com KCl tem um efeito estabilizador em alguns complexos Glicerol (1–10%): estabiliza as proteínas |
| Ligado à membrana | Buffer RIPA | Veja acima |
| Mitocôndria | Buffer RIPA | |
| Nuclear | Buffer RIPA | Veja acima; note: O NP-40 e o Triton X-100 não conseguem lisar membranas nucleares. |
| Localização da proteína | Buffer |
A percentagem de gel necessária depende do tamanho da proteína de interesse:
| Tamanho da proteína | Porcentagem de gel |
| 4–40 kDa | 20% |
| 12–45 kDa | 15% |
| 10–70 kDa | 12% |
| 15–100 kDa | 10% |
| 25–100 kDa | 8% |
| 60-210 kDa | 5% |
Também podem ser utilizados géis gradientes.
| Tamanho da proteína | Porcentagem de gel |
| 5-200 kDa | 4-12 gradient |
| 4-200 kDa | 4-20 gradient |
| 3.5-110 kDa | 10-20 gradient |
Sistemas de buffer contínuos versus descontínuos
Os sistemas de separação eletroforética podem ser amplamente classificados em sistemas tampão contínuos e discontínuos (também chamados multifásicos ou isotacophoréticos), cada um caracterizado por configurações tampão e dinâmicas de separação distintas.
1. Sistemas de tampão contínuo:
-
Os mesmos iões tampão e pH em toda a amostra, gel e reservatórios.
-
Gel feito com uma única concentração de acrilamida.
-
As proteínas separam-se diretamente no gel sem empilhar.
-
Ponto principal: A resolução depende muito de cargas de amostras pequenas e concentradas.
2. Sistemas tampão descontínuos (multifásicos):
-
Dois tampões diferentes com pH e força iónica distintos:
-
Gel de empilhamento: pH ~6,8, baixo Tris–HCl (0,125 M), poros grandes.
-
Gel de resolução: pH ~8,8, alto Tris–HCl (0,375 M), poros pequenos.
-
-
Tampão de corrida (por exemplo, Tris-glicina) introduzido acima do gel de empilhamento.
3. Mecanismo de separação:
-
Os iões Cl⁻ atuam como iões principais; o glicinato como iões secundários.
-
As proteínas concentram-se em bandas estreitas no gel de empilhamento («efeito de empilhamento»).
-
No gel de resolução, as proteínas separam-se por peso molecular à medida que se tornam os principais iões.
4. Vantagens dos sistemas descontínuos:
-
Permite o carregamento de volumes maiores de amostras diluídas.
-
Resolução superior devido ao empilhamento e aos diferentes tamanhos dos poros do gel.
-
Separação mais eficaz com base no peso molecular.
Transferência de proteínas
Após a eletroforese, as proteínas são transferidas de um gel de poliacrilamida para uma membrana. Esta etapa é crucial para imobilizar as proteínas para posterior deteção e análise (por exemplo, western blotting).
1. Membranas utilizadas para transferência de proteínas
Estão disponíveis diferentes membranas, cada uma com vantagens específicas:
-
A nitrocelulose é a membrana mais utilizada. Possui alta afinidade de ligação com proteínas, pode imobilizar tanto proteínas quanto glicoproteínas e é compatível com métodos de detecção por quimioluminescência, cromogênica e fluorescente. No entanto, é frágil, pois é derivada da celulose tratada com ácido nítrico.
-
PVDF (difluoreto de polivinilideno) oferece melhor resistência mecânica do que a nitrocelulose, tornando-o ideal para remoção e reprovação ou para sequenciamento de proteínas. No entanto, o PVDF pode, por vezes, produzir uma coloração de fundo mais elevada, exigindo otimização adicional.
2. Métodos para transferência de proteínas
As proteínas podem ser transferidas do gel para a membrana utilizando vários métodos:
-
A transferência capilar depende da ação capilar passiva.
-
A transferência por difusão permite que as proteínas se difundam lentamente na membrana.
-
Electroblotting é o método mais rápido e completo para transferência.
Entre estes, o electroblotting é o método preferido pela sua rapidez e eficiência.
Tipos de eletroblotting
Existem três tipos de electroblotting com base no método de utilização do tampão:
-
A electroblotting a seco utiliza componentes secos sem adição de líquido.
-
Aelectroblotting semi-seca utiliza papéis de filtro embebidos em tampão de transferência entre a membrana e o gel.
-
Aelectroblotting húmida imerge todo o conjunto gel-membrana num tampão de transferência, o que é particularmente recomendado para a transferência de proteínas de grande dimensão.
1. Princípios da eletroblotting
Durante a eletroblotting:
-
Um campo elétrico é aplicado perpendicularmente ao gel.
-
As proteínas, por serem carregadas, migram do gel para a membrana.
-
O gel deve estar voltado para o eletrodo negativo (cátodo), enquanto a membrana deve estar voltada para o eletrodo positivo (ânodo).
-
Para garantir uma transferência adequada, o gel e a membrana são firmemente unidos, com papéis de filtro e almofadas de fibra colocadas em ambos os lados para evitar bolhas, que podem causar uma transferência irregular.
a.Buffers de transferência
Os tampões de transferência ajudam a manter a estrutura das proteínas e melhoram o movimento durante a transferência. Os tampões comuns incluem:
-
O tampão Towbin (25 mM Tris, 192 mM glicina, 20% metanol, pH 8,3) é amplamente utilizado para transferência húmida.
-
O tampão Bjerrum Schafer-Nielsen (48 mM Tris, 39 mM glicina, pH 9,2, 20% metanol) é preferível para transferência semi-seca.
-
O tampão CAPS (60 mM Tris e 40 mM CAPS) é utilizado especialmente para proteínas com pontos isoelétricos elevados.
O metanol no tampão ajuda a evitar o inchaço do gel e melhora a ligação das proteínas à membrana. No entanto, em alguns casos, tampões sem metanol ainda podem produzir sinais de detecção fortes.
A função dos componentes do tampão de transferência é descrita na Tabela 2.
Tabela 2. Composição do tampão de execução e transferência e função de cada componente
| Componente potencial | Função |
| Buffers em execução | |
| Acetate | Boa separação na gama de massa mais elevada (100–500 kDa) |
| Glycine | Forneça iões de glicinato para formar a extremidade final durante a eletroforese em gel de estaca. |
| MES | Necessário para manter o pH relativamente constante. Fornece iões MES para formar a extremidade final durante a eletroforese. O MES resulta numa separação maior na gama de massa mais baixa (<50 kDa). O tampão MES SDS é mais rápido do que o tampão MOPS SDS devido ao pKa mais baixo do MES em comparação com o MOPS. |
| MOPS | Necessário para manter um pH relativamente constante. Fornece iões MOPS para formar a extremidade final durante a eletroforese. O MOPS proporciona uma melhor separação na gama de massa média a elevada. A diferença no movimento dos iões MES e MOPS afeta o empilhamento, resultando numa diferença na gama de separação das proteínas entre os tampões MES e MOPS. |
| SDS | Ajuda a manter as proteínas com uma carga líquida negativa |
| Tricine | Necessário para manter o pH relativamente constante. A tricina substitui a glicina no tampão de corrida, fornecendo iões de tricina para formar a extremidade final durante a eletroforese. A tricina resulta numa empilhagem e resolução mais eficientes das proteínas de baixo peso molecular. Adequado para separar proteínas de baixo peso molecular (0,5–50 kDa). |
| Tris-HCl | Necessário para manter o pH relativamente constante. Fornece iões de cloreto que formam a extremidade principal durante a eletroforese em gel de estaca. Os iões de hidrogénio conduzem a eletricidade. |
| Transfer buffers | |
| Tris-HCl | Necessário para manter o pH relativamente constante |
| Glycine | Na ausência de metanol, ajuda a evitar que o gel inche. |
| Methanol | Impede que o gel inche durante a transferência e melhora a ligação das proteínas à nitrocelulose. Remove o SDS das proteínas, permitindo uma melhor ligação às membranas de nitrocelulose. Alguns laboratórios já não incluem metanol nos tampões de transferência, uma vez que os resultados sem metanol não foram significativamente diferentes dos resultados com metanol. Alguns protocolos utilizam etanol a 10% em vez de metanol a 20%. |
| SDS | O SDS (até 0,1%) no tampão de transferência aumenta a eficiência da transferência de proteínas, especialmente para proteínas grandes, mas pode reduzir a quantidade de proteína que se liga à membrana. Em membranas de nitrocelulose com poros maiores (0,45 μm), pequenas proteínas desnaturadas pelo SDS podem atravessar a membrana. |
| CAPS | Necessário para manter um pH relativamente constante. Recomendado para transferência de proteínas de alto peso molecular (>150 kDa) |
| CAPS: (Ciclohexilamino)-L-propanossulfónico; MES: 2-(N-morfolino) etanossulfónico; MOPS: 3-(N-morfolino) propanossulfónico; SDS: Dodecilsulfato de sódio; Tris-HCl: Cloridrato de tris. |
|
b.Considerações importantes
-
As proteínas com peso molecular muito baixo (<10 kDa) podem ser transferidas de forma ineficiente.
-
Se o pH do tampão de transferência for inferior ao ponto isoelétrico da proteína, as proteínas podem migrar para trás.
-
Tampões especializados (como tampões à base de CAPS) são frequentemente necessários para transferir eficientemente proteínas com um ponto isoelétrico elevado.
Etapa de bloqueio na transferência Western
Após a transferência bem-sucedida da proteína para a membrana, o bloqueio é uma etapa crítica para garantir a especificidade e a clareza da imunodetecção subsequente.

1.Objetivo e importância
-
O bloqueio impede a ligação não específica de anticorpos à membrana, minimizando assim o ruído de fundo e eliminando sinais falsos positivos.
-
Aumenta a precisão e a fiabilidade da deteção de proteínas durante a transferência Western.
2. Procedimento de bloqueio
-
A membrana é incubada durante 1 hora à temperatura ambiente numa solução de bloqueio contendo:
-
Albumina sérica bovina (BSA) ou leite em pó desnatado, diluído em:
-
Solução salina tamponada com Tris com 0,1% de Tween 20 (TBST) ou
-
Solução salina tamponada com fosfato com 0,1% de Tween 20 (PBST).
-
-
- As proteínas presentes na BSA e no leite (principalmente caseína e proteínas do soro do leite) ligam-se a locais não ocupados na membrana, impedindo assim interações não específicas entre a membrana e os primários ou secundários anticorpos.
3. Características da membrana e função do Tween 20
-
As membranas de nitrocelulose e PVDF apresentam elevadas afinidades de ligação às proteínas, predominantemente através de interações hidrofóbicas.
-
Embora a BSA e as proteínas do leite possam associar-se fracamente às proteínas transferidas, a adição de Tween 20 no tampão de bloqueio reduz essas interações não específicas, garantindo um bloqueio mais eficaz.
Incubação e deteção de anticorpos em Western blotting
1. O papel da incubação de anticorpos na Western blotting
Após bloquear a membrana para impedir a ligação não específica, o western blotting passa para a etapa crucial da incubação do anticorpo. Primeiro, a membrana é incubada com um anticorpo primário que se liga especificamente à proteína alvo. Após lavar os anticorpos não ligados com tampões como TBST ou PBST, é introduzido um anticorpo secundário. Este anticorpo secundário reconhece o anticorpo primário e é normalmente conjugado a uma enzima como HRP (peroxidase de rábano) ou fosfatase alcalina (AP), permitindo a deteção subsequente do sinal.
-
Anticorpos primários: Essenciais para a ligação específica e seletiva às proteínas-alvo.
-
Anticorpos secundários: Permitem a deteção e amplificação do sinal.
Dica: É importante otimizar a concentração de anticorpos para maximizar a sensibilidade e reduzir o ruído de fundo. O nosso suporte técnico pode ajudá-lo com a otimização do protocolo.
Os passos de lavagem são essenciais para remover anticorpos não ligados. No entanto, uma lavagem excessiva (>20 minutos) pode enfraquecer o sinal — por isso, é fundamental encontrar o equilíbrio certo!
2. Métodos de deteção de sinais
Depois que a proteína alvo é ligada aos anticorpos marcados, o sinal deve ser detectado. Existem vários métodos disponíveis:
a. Quimioluminescência (ECL)
A quimioluminescência melhorada (ECL) é o método de deteção mais popular. Utilizando anticorpos secundários conjugados com HRP e substratos ECL, é gerada luz que é capturada em filme ou com um dispositivo de imagem digital.
-
Altamente sensível
-
Ampla gama dinâmica
-
Ideal para detetar proteínas de baixa abundância
Produtos recomendados:
Anticorpos secundários conjugados com HRP
Kits de substratos ECL
Kits de detecção ECL
b. Detecção por fluorescência
Os anticorpos secundários conjugados com corante fluorescente permitem a detecção multiplex, o que significa que é possível visualizar várias proteínas ao mesmo tempo utilizando diferentes fluoróforos.
-
Adequado para deteção simultânea de vários alvos
-
Excelente gama dinâmica e quantificação
-
Ideal para utilização com fluoróforos na gama do infravermelho
Produtos recomendados:
Anticorpos secundários (gama visível e infravermelha)
Sistemas de imagem fluorescente
Anticorpos fluorescentes
c. Detecção cromogénica
Os métodos cromogénicos utilizam reações enzimáticas para gerar um precipitado colorido na membrana, visível a olho nu.
- Ideal quando o equipamento de imagem é limitado.
-
Fácil visualização
-
Não é necessário equipamento especial
-
Ideal para laboratórios educacionais e investigação básica
Produtos recomendados:
Kits de substratos cromogénicos (e.g., BCIP/NBT)
d. Detecção radioativa (menos comum atualmente)
As sondas radioativas eram amplamente utilizadas para western blotting, mas agora foram substituídas por métodos mais seguros e igualmente sensíveis, como ECL e fluorescência. A seleção do método de deteção adequado é fundamental para o sucesso do seu Western blot e depende da sensibilidade, especificidade e tipo de análise necessária:
-
Sensibilidade máxima? → Escolha a deteção Quimioluminescência Avançada (ECL).
A ECL oferece alta sensibilidade e ampla gama dinâmica, tornando-a ideal para detectar proteínas de baixa abundância. A intensidade do sinal pode ser capturada em filmes de raios X ou sistemas de imagem digital. -
Análise multiplex (detecção simultânea de vários alvos)? → Escolha detecção baseada em fluorescência.
Os anticorpos secundários fluorescentes permitem a detecção simultânea de múltiplas proteínas sem a necessidade de remover e reprovar as membranas. Os sinais são altamente estáveis, quantitativos e ideais para estudos comparativos. -
Visualização simples e rápida? → Escolha a detecção cromogénica.
Os sistemas cromogénicos utilizam reações enzimático-substrato para produzir um precipitado colorido diretamente na membrana. Embora menos sensíveis do que o ECL ou a fluorescência, são fáceis de usar e não requerem equipamento de imagem especializado.
Além disso, a escolha dos anticorpos primários e secundários, bem como dos substratos ou reagentes de detecção, deve ser compatível com o método de detecção selecionado. O emparelhamento adequado é essencial para obter sinais específicos e fortes, minimizando o ruído de fundo.