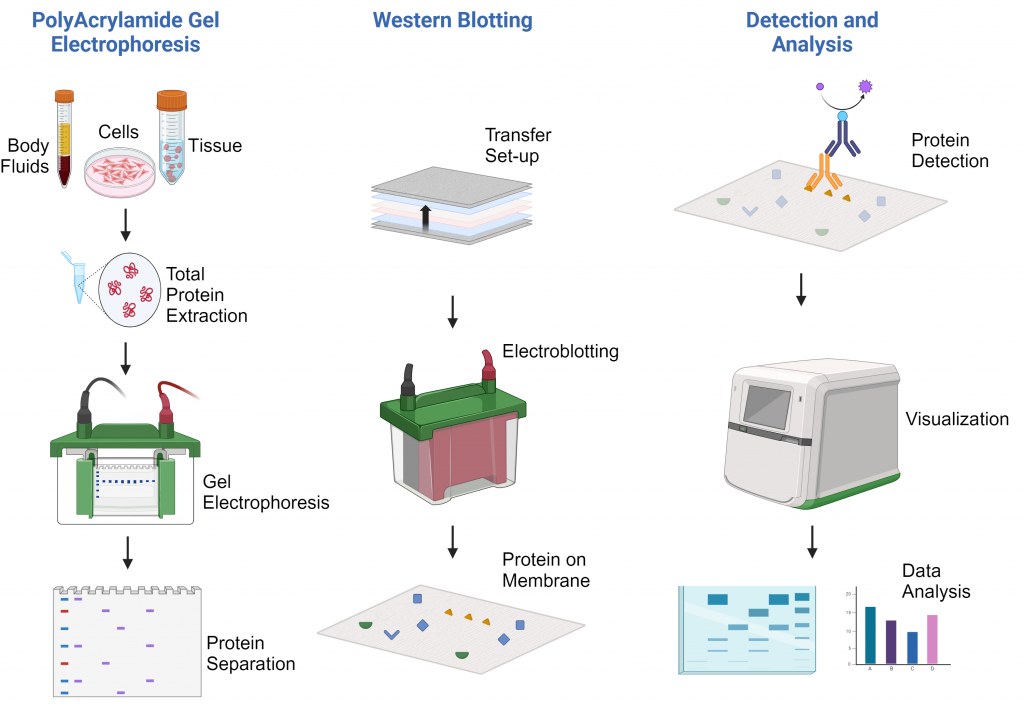
Western Blot Protocol

Principle of the Technique
The principle of Western blotting involves several critical steps:
-
Protein Separation: Proteins are separated by size through SDS-PAGE.
-
Transfer: Separated proteins are transferred onto a solid membrane (nitrocellulose or PVDF).
-
Blocking: Membrane surfaces are blocked to prevent non-specific binding.
-
Detection: Specific primary antibodies recognize target proteins, followed by labeled secondary antibodies.
-
Visualization: Protein-antibody complexes are visualized via chemiluminescent, fluorescent, or colorimetric reactions.
The technique’s success relies heavily on antibody specificity and the integrity of the sample preparation.
The first important step that should not be overlooked when doing western blotting is sample preparation. To get reproducible results, efficient protein extraction and purification should be carried out using a suitable homogenization method that can efficiently release the intracellular contents of the cell through the rupture of cell membranes.
Protein Sample Isolation from Different Types of Samples
Protein extraction and isolation are critical first steps in a wide range of biochemical and molecular biology techniques, including Western blotting, mass spectrometry, enzyme assays, and immunoprecipitation. The success of downstream applications heavily depends on the quality, integrity, and representativeness of the isolated protein samples.
Biological samples such as cultured cells, tissues, bacteria, yeast, and plant material each present unique challenges for protein extraction. Therefore, choosing an appropriate isolation strategy is essential to maximize yield, preserve protein functionality, and minimize degradation or modification.
1. Homogenization
-
Choose a homogenization method based on sample type:
-
Ultrasonication (for delicate samples)
-
French press (for bacteria, yeast)
-
Glass-bead milling (for tough cells, fungi)
-
Dounce/Potter-Elvehjem homogenizer (for soft tissues)
-
Manual grinding with mortar/pestle (for frozen tissues)
-
-
Goal: Efficient cell lysis without protein degradation.
2. Sample Handling Post-Treatment
-
After xenobiotic exposure or genetic manipulation (e.g., siRNA knockdown):
-
Immediately freeze samples in liquid nitrogen
-
Lysate immediately to prevent endogenous protease activity.
-
-
Always keep samples cold during processing.
3. Freeze/Thaw Cycle Management
-
Avoid multiple freeze/thaw cycles to protect protein integrity.
-
Aliquot samples to avoid unnecessary thawing.
-
4. Lysis Buffer Selection
a.Match buffer to target protein location:
-
Cytosolic proteins → Mild lysis buffer (e.g., NP-40, Triton X-100)
-
Membrane-bound proteins → Stronger buffers (e.g., RIPA)
-
Nuclear/mitochondrial proteins → Specialized buffers ( e.g,Hypotonic Buffer, Drosophila Homogenization Buffer)
b.Match buffer to antibody sensitivity:
-
Native protein detection → Mild/ nonionic detergent or no detergent
-
Denatured protein detection → Strong/denaturing conditions
-
Include protease and phosphatase inhibitors in all lysis buffers.
c.Some Common Ingredients and Why They’re Used:
-
HEPES: Maintains pH stability.
-
Sucrose / Mannitol: Osmotic stabilizers to prevent organelle bursting.
-
EDTA / EGTA: Chelates divalent cations to inhibit metalloproteases and prevent degradation.
-
Protease and phosphatase inhibitors: Protect proteins from enzymatic degradation.
5. Critical Notes on Sample Fractionation
a.Beware of protein loss during centrifugation:
-
Test if the target protein is retained in the pellet (debris).
-
Example: Up to 50% of myosin and ~66% of CSQ2 were lost into debris if not checked.
b.Always verify whether the protein of interest is:
-
Soluble (cytoplasmic)
-
Insoluble (cytoskeletal)
-
Membrane-bound (organellar)
Electrophoretic separation
Following protein extraction and quantification, the next critical phase in proteomic analysis is electrophoretic separation. Gel electrophoresis remains a foundational technique for enhancing selectivity and sensitivity in proteome research, allowing resolution of complex protein mixtures based on physicochemical properties.

Various gel matrices—including agarose, starch, and polyacrylamide—have been developed for electrophoretic applications. Among these, polyacrylamide is the matrix of choice for protein analysis due to its mechanical stability, chemical inertness, and the ability to precisely regulate polymerization with bisacrylamide. A commonly employed acrylamide:bisacrylamide ratio of 37.5:1 enables the generation of robust gels with reproducible pore sizes, suitable for high-resolution protein fractionation.
Two principal forms of polyacrylamide gel electrophoresis (PAGE) are widely utilized: native PAGE and denaturing SDS-PAGE.
-
Native PAGE preserves the tertiary and quaternary structures of proteins, enabling their separation based on a combination of net charge, hydrodynamic size, and conformation. In alkaline buffer systems, most proteins acquire a net negative charge, allowing migration toward the anode without denaturation. A major advantage of native PAGE is the retention of enzymatic activity and native protein–protein interactions during separation.
-
SDS-PAGE (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) is the most commonly employed method for separating denatured proteins based solely on molecular weight. The anionic detergent SDS binds uniformly along the polypeptide backbone, imparting a consistent negative charge proportional to protein length. Under denaturing conditions (typically with heating in 0.1% SDS), proteins are linearized and adopt an extended rod-like shape, enabling size-dependent migration through the polyacrylamide matrix. Smaller proteins traverse the gel more rapidly than larger counterparts. Furthermore, SDS-PAGE benefits from isotachophoresis principles, concentrating protein samples into tight, defined bands and thereby improving resolution (≥ tenfold concentration observed in controlled laboratory settings).
Attention to sample preparation, buffer composition, and gel characteristics is essential for achieving optimal separation and reproducibility. The components of typical sample buffers and their respective functions are summarized in Table 1.
Table 1. Commonly used sample preparation buffers. Protein location Buffer Purpose
| Protein location | Buffer | Purpose of components (typical concentrations shown in brackets) |
| Whole cell | Nondenaturing, nonionic buffer: NP-40 buffer: 50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40 or Triton X-100 |
Tris-HCl (10–50 mM): buffering of solution (helps maintain stability of proteins) NaCl (50–150 mM): reduces nonspecific binding, maintains ionic strength NP-40/Triton X-100 (0.1–1%): nonionic detergents, solubilizes cytosolic and membrane proteins without denaturing them, permeabilizes membranes and increases solubility of proteins preventing aggregation |
| Denaturing buffer: RIPA buffer: 150 mM NaCl, 1% NP-40 or Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0 |
Sodium deoxycholate (0.1–0.5%): ionic detergent, lyse cells and solubilize cellular and membrane components SDS (0.1–1%): ionic detergent, causes membrane disruption and linearizes proteins by binding to them |
|
| Cytoplasmic (soluble) |
20 mM Tris-HCl, pH 7.5 or NP-40 buffer | See above |
| Cytoplasmic (cytoskeletal-bound) |
PIPES-Triton buffer: 10 mM PIPES, 50 mM KCl, 1% Triton X-100, 10 mM EGTA, 3 mM MgCl2, 2 M glycerol |
PIPES (10 M): buffering of solution KCl (50–150 mM): reduces nonspecific binding, maintains ionic strength EGTA (1–3 mM): inhibition of Ca2+ metalloproteases MgCl2 (1–5 mM): reduces nonspecific binding, together with KCl has a stabilizing effect on some complexes Glycerol (1–10%): stabilizes proteins |
| Membrane-bound | RIPA buffer | See above |
| Mitochondria | RIPA buffer | See above |
| Nuclear | RIPA buffer | See above; note: NP-40 and Triton X-100 cannot lyse nuclear membranes |
| Protein location | Buffer |
The gel percentage required is dependent on the size of your protein of interest:
| Protein size | Gel percentage |
| 4–40 kDa | 20% |
| 12–45 kDa | 15% |
| 10–70 kDa | 12% |
| 15–100 kDa | 10% |
| 25–100 kDa | 8% |
| 60-210 kDa | 5% |
Gradient gels can also be used.
| Protein size | Gel percentage |
| 5-200 kDa | 4-12 gradient |
| 4-200 kDa | 4-20 gradient |
| 3.5-110 kDa | 10-20 gradient |
Continuous versus Discontinuous Buffer Systems
Electrophoretic separation systems can broadly be classified into continuous and discontinuous (also called multiphasic or isotachophoretic) buffer systems, each characterized by distinct buffer configurations and separation dynamics.
1. Continuous Buffer Systems:
-
Same buffer ions and pH across sample, gel, and reservoirs.
-
Gel made of a single acrylamide concentration.
-
Proteins separate directly in the gel without stacking.
-
Key point: Resolution depends heavily on small, concentrated sample loads.
2. Discontinuous (Multiphasic) Buffer Systems:
-
Two different buffers with distinct pH and ionic strength:
-
Stacking gel: pH ~6.8, low Tris–HCl (0.125 M), large pores.
-
Resolving gel: pH ~8.8, high Tris–HCl (0.375 M), small pores.
-
-
Running buffer (e.g., Tris-glycine) introduced above the stacking gel.
3. Separation mechanism:
-
Cl⁻ ions act as leading ions; glycinate as trailing ions.
-
Proteins concentrate into tight bands in the stacking gel ("stacking effect").
-
In the resolving gel, proteins separate by molecular weight as they become the main ions.
4. Advantages of Discontinuous Systems:
-
Allows loading of larger volumes of dilute samples.
-
Superior resolution due to stacking and different gel pore sizes.
-
More effective separation based on molecular weight.
Protein Transfer
After electrophoresis, proteins are transferred from a polyacrylamide gel onto a membrane. This step is crucial to immobilize proteins for further detection and analysis (e.g., western blotting).
1. Membranes Used for Protein Transfer
Different membranes are available, each with specific advantages:
-
Nitrocellulose is the most commonly used membrane. It has a high protein-binding affinity, can immobilize both proteins and glycoproteins, and is compatible with chemiluminescence, chromogenic, and fluorescent detection methods. However, it is brittle because it is derived from cellulose treated with nitric acid.
-
PVDF (Polyvinylidene difluoride) offers better mechanical strength than nitrocellulose, making it ideal for stripping and reprobing or for sequencing proteins. However, PVDF can sometimes produce higher background staining, requiring additional optimization.
2. Methods for Protein Transfer
Proteins can be transferred from the gel to the membrane using several methods:
-
Capillary transfer relies on passive capillary action.
-
Diffusion transfer allows proteins to slowly diffuse onto the membrane.
-
Electroblotting is the fastest and most complete method for transfer.
Among these, electroblotting is the preferred method for its speed and efficiency.
Types of Electroblotting
There are three types of electroblotting based on the method of buffer usage:
-
Dry electroblotting uses dry components without added liquid.
-
Semi-dry electroblotting uses transfer buffer-soaked filter papers between the membrane and gel.
-
Wet electroblotting immerses the entire gel-membrane sandwich in a transfer buffer, which is particularly recommended for transferring large proteins.
1. Principles of Electroblotting
During electroblotting:
-
An electric field is applied perpendicular to the gel.
-
Proteins, being charged, migrate out of the gel onto the membrane.
-
The gel must face the negative electrode (cathode) while the membrane faces the positive electrode (anode).
-
To ensure proper transfer, the gel and membrane are sandwiched together tightly, with filter papers and fiber pads placed on either side to prevent bubbles, which could cause uneven transfer.
a.Transfer Buffers
Transfer buffers help maintain protein structure and enhance movement during the transfer. Common buffers include:
-
Towbin buffer (25 mM Tris, 192 mM glycine, 20% methanol, pH 8.3) is widely used for wet transfer.
-
Bjerrum Schafer-Nielsen buffer (48 mM Tris, 39 mM glycine, pH 9.2, 20% methanol) is preferred for semi-dry transfer.
-
CAPS buffer (60 mM Tris and 40 mM CAPS) is used especially for proteins with high isoelectric points.
Methanol in the buffer helps prevent gel swelling and improves protein binding to the membrane. However, in some cases, methanol-free buffers can still produce strong detection signals.
The role of the components of the transfer buffer are described in Table 2.
Table 2. Running and transfer buffer composition and role of each component
| Potential component | Role |
| Running buffers | |
| Acetate | Good separation in higher mass range (100–500 kDa) |
| Glycine | Provide glycinate ions to form trailing end during electrophoresis in staking gel |
| MES | Needed to maintain relatively constant pH. Provides MES ions to form trailing end during electrophoresis. MES results in a larger separation at lower mass range (<50 kDa). MES SDS buffer is faster than MOPS SDS buffer due to MES’s lower pKa compared with MOPS |
| MOPS | Needed to maintain relatively constant pH. Provides MOPS ions to form trailing end during electrophoresis. MOPS gives better separation at middle to higher mass range. The difference in MES and MOPS ion movement impacts stacking resulting in a difference in protein separation range between MES and MOPS buffers |
| SDS | Helps keeps proteins in a net-negative charge |
| Tricine | Needed to maintain relatively constant pH. Tricine substitutes for glycine in running buffer, providing tricine ions to form trailing end during electrophoresis. Tricine results in more efficient stacking and resolving of low molecular weight proteins. Good for separating low molecular weight proteins (0.5–50 kDa) |
| Tris-HCl | Needed to maintain relatively constant pH. Provides chloride ions that form leading end during electrophoresis in staking gel. Hydrogen ions conduct electricity |
| Transfer buffers | |
| Tris-HCl | Needed to maintain relatively constant pH |
| Glycine | In the absence of methanol, helps prevent gel from swelling |
| Methanol | Prevents gel from swelling during transfer and improves protein binding to nitrocellulose. Removes SDS from proteins allowing better binding to nitrocellulose membranes. Some laboratories no longer include methanol in transfer buffers as results without methanol were not significantly different from those with methanol. Some protocols use 10% ethanol instead of 20% methanol |
| SDS | SDS (up to 0.1%) in transfer buffer increases protein transfer efficiency, especially for large proteins, but may reduce amount of protein binding to membrane. In nitrocellulose membranes with larger pore sizes (0.45 μm), small SDS denatured proteins may go through the membrane |
| CAPS | Needed to maintain relatively constant pH. Recommended for transfer of high molecular weight proteins (>150 kDa) |
| CAPS: (Cyclohexylamino)-L-propanesulfonic acid; MES: 2-(N-morpholino) ethanesulfonic acid; MOPS: 3-(N-morpholino) propanesulfonic acid; SDS: Sodium Dodecyl Sulfate; Tris-HCl: Tris hydrochloride. |
|
b.Important Considerations
-
Proteins with very low molecular weight (<10 kDa) may transfer inefficiently.
-
If the transfer buffer pH is lower than the protein's isoelectric point, proteins may migrate backward.
-
Specialized buffers (such as CAPS-based buffers) are often needed for efficiently transferring proteins with a high isoelectric point.
Blocking Step in Western Blotting
Following successful protein transfer to the membrane, blocking is a critical step to ensure the specificity and clarity of subsequent immunodetection.

1.Purpose and Importance
-
Blocking prevents nonspecific binding of antibodies to the membrane, thereby minimizing background noise and eliminating false-positive signals.
-
It enhances the accuracy and reliability of protein detection during western blotting.
2. Blocking Procedure
-
The membrane is incubated for 1 hour at room temperature in a blocking solution containing:
-
Bovine Serum Albumin (BSA) or nonfat dry milk, diluted in:
-
Tris-buffered saline with 0.1% Tween 20 (TBST), or
-
Phosphate-buffered saline with 0.1% Tween 20 (PBST).
-
-
-
The proteins present in BSA and milk (primarily casein and whey proteins) bind to unoccupied sites on the membrane, thereby preventing nonspecific interactions between the membrane and the primary or secondary antibodies.
3. Membrane Characteristics and Role of Tween 20
-
Nitrocellulose and PVDF membranes exhibit high protein-binding affinities predominantly through hydrophobic interactions.
-
While BSA and milk proteins may weakly associate with transferred proteins, the addition of Tween 20 in the blocking buffer reduces these nonspecific interactions, ensuring more effective blocking.
Antibody Incubation and Detection in Western Blotting
1. The Role of Antibody Incubation in Western Blotting
After blocking the membrane to prevent nonspecific binding, western blotting moves to the crucial step of antibody incubation. First, the membrane is incubated with a primary antibody that specifically binds the target protein. After washing away unbound antibodies with buffers like TBST or PBST, a secondary antibody is introduced. This secondary antibody recognizes the primary antibody and is typically conjugated to an enzyme such as HRP (Horseradish Peroxidase) or Alkaline Phosphatase (AP), enabling subsequent signal detection.
-
Primary Antibodies: Essential for specific and selective binding to target proteins.
-
Secondary Antibodies: Enable detection and amplification of the signal.
Tip: It's important to optimize antibody concentration to maximize sensitivity and reduce background. Our technical support can help you with protocol optimization.
Washing steps are critical to remove unbound antibodies. However, excessive washing (>20 minutes) may weaken the signal — so balanced timing is key!
2. Signal Detection Methods
Once the target protein is bound by labeled antibodies, the signal must be detected. Several methods are available:
a. Chemiluminescence (ECL)
Enhanced chemiluminescence (ECL) is the most popular detection method. Using HRP-conjugated secondary antibodies and ECL substrates, light is generated and captured either on film or with a digital imager.
-
Highly sensitive
-
Wide dynamic range
-
Ideal for detecting low-abundance proteins
Recommended Products:
HRP-conjugated Secondary Antibodies
ECL Substrate Kits
ECL Detection Kits
b. Fluorescence Detection
Fluorescent dye-conjugated secondary antibodies allow multiplex detection — meaning you can visualize multiple proteins at once using different fluorophores.
-
Suitable for simultaneous multi-target detection
-
Excellent dynamic range and quantitation
-
Best when using infrared range fluorophores
Recommended Products:
Secondary Antibodies (Visible and Infrared Range)
Fluorescent Imaging Systems
Fluorescent Antibodies
c. Chromogenic Detection
Chromogenic methods use enzyme reactions to generate a colored precipitate on the membrane, visible to the naked eye.
- Ideal when imaging equipment is limited.
-
Easy visualization
-
No need for special equipment
-
Ideal for educational labs and basic research
Recommended Products:
Chromogenic Substrate Kits (e.g., BCIP/NBT)
d. Radioactive Detection (Less Common Today)
Radioactive probes were once widely used for western blotting but are now largely replaced by safer, equally sensitive methods like ECL and fluorescence. Selecting the appropriate detection method is critical for the success of your Western blot and depends on the sensitivity, specificity, and type of analysis you require:
-
Maximum sensitivity? → Choose Enhanced Chemiluminescence (ECL) detection.
ECL offers high sensitivity and broad dynamic range, making it ideal for detecting low-abundance proteins. Signal intensity can be captured on X-ray films or digital imaging systems. -
Multiplex analysis (detecting multiple targets simultaneously)? → Choose fluorescence-based detection.
Fluorescent secondary antibodies allow simultaneous detection of multiple proteins without the need to strip and reprobe membranes. Signals are highly stable, quantitative, and ideal for comparative studies. -
Simple, quick visualization? → Choose chromogenic detection.
Chromogenic systems use enzyme-substrate reactions to produce a colored precipitate directly on the membrane. While less sensitive than ECL or fluorescence, they are easy to use and do not require specialized imaging equipment.
Additionally, the choice of primary and secondary antibodies, as well as substrates or detection reagents, must be compatible with your selected detection method. Proper pairing is essential to achieve specific and strong signals while minimizing background noise.