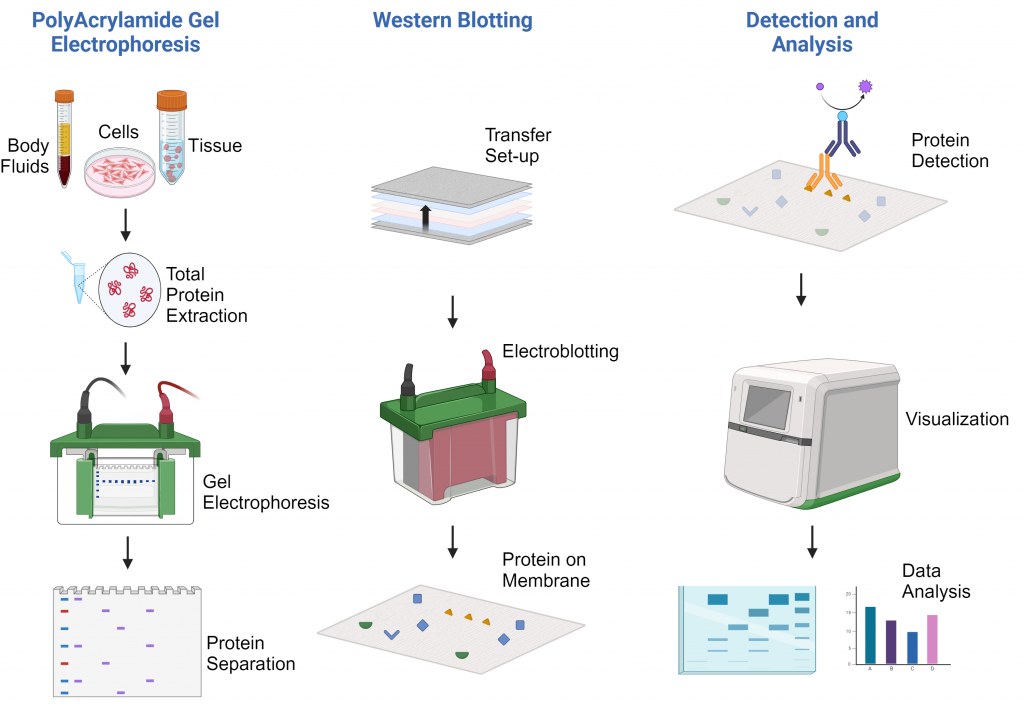
Protocollo Western Blot

Principio della Tecnica
Il principio del Western blotting si basa su diverse fasi fondamentali:
-
Separazione delle proteine: Le proteine vengono separate in base alla dimensione mediante SDS-PAGE.
-
Trasferimento: Le proteine separate vengono trasferite su una membrana solida (nitrocellulosa o PVDF).
-
Bloccaggio: Le superfici della membrana vengono bloccate per prevenire legami aspecifici.
-
Rivelazione: Anticorpi primari specifici riconoscono le proteine target, seguiti da anticorpi secondari marcati.
-
Visualizzazione: I complessi proteina-anticorpo vengono visualizzati tramite reazioni chemioluminescenti, fluorescenti o colorimetriche.
Il successo della tecnica dipende fortemente dalla specificità degli anticorpi e dall’integrità della preparazione del campione.
Il primo passaggio cruciale da non trascurare nel Western blotting è la preparazione del campione. Per ottenere risultati riproducibili, è necessario eseguire un'efficiente estrazione e purificazione proteica utilizzando un metodo di omogeneizzazione adeguato, capace di rilasciare efficacemente il contenuto intracellulare tramite la rottura delle membrane cellulari.
Isolamento del Campione Proteico da Diverse Tipologie di Campioni
L’estrazione e l’isolamento delle proteine rappresentano fasi critiche in numerose tecniche biochimiche e di biologia molecolare, tra cui Western blotting, spettrometria di massa, saggi enzimatici e immunoprecipitazione. Il successo delle applicazioni successive dipende fortemente dalla qualità, integrità e rappresentatività dei campioni proteici isolati.
I campioni biologici come cellule in coltura, tessuti, batteri, lieviti e materiale vegetale presentano ciascuno sfide specifiche per l’estrazione proteica. Pertanto, la scelta di una strategia di isolamento adeguata è essenziale per massimizzare la resa, preservare la funzionalità delle proteine e minimizzare la degradazione o le modificazioni.
1. Omogeneizzazione
-
Scegliere un metodo di omogeneizzazione in base al tipo di campione:
-
Ultrasuonazione (per campioni delicati)
-
Pressa francese (per batteri, lieviti)
-
Frantumazione con sfere di vetro (per cellule resistenti, funghi)
-
Omogeneizzatore Dounce/Potter-Elvehjem (per tessuti molli)
-
Macinazione manuale con mortaio e pestello (per tessuti congelati)
-
-
Obiettivo: Lisi cellulare efficiente senza degradazione delle proteine.
2. Gestione del Campione dopo il Trattamento
-
Dopo esposizione a xenobiotici o manipolazione genetica (es. silenziamento con siRNA):
-
Congelare immediatamente i campioni in azoto liquido
-
Lizzare immediatamente per prevenire l’attività delle proteasi endogene.
-
-
Mantenere sempre i campioni a bassa temperatura durante l’elaborazione.
3. Gestione dei Cicli di Congelamento/Scongelamento
-
Evitare cicli ripetuti di congelamento/scongelamento per proteggere l’integrità proteica.
-
Suddividere i campioni in aliquote per evitare scongelamenti non necessari.
-
4. Selezione del Tampone di Lisi
a. Abbinare il tampone alla localizzazione della proteina target:
-
Proteine citosoliche → Tampone di lisi blando (es. NP-40, Triton X-100)
-
Proteine di membrana → Tamponi più forti (es. RIPA)
-
Proteine nucleari/mitocondriali → Tamponi specializzati (es. Tampone ipotonico, Tampone di omogeneizzazione per Drosophila)
b. Abbinare il tampone alla sensibilità dell’anticorpo:
-
Rilevazione di proteine native → Detergente non ionico blando o assenza di detergente
-
Rilevazione di proteine denaturate → Condizioni forti/denaturanti
-
Includere inibitori di proteasi e fosfatasi in tutti i tamponi di lisi.
c. Alcuni Ingredienti Comuni e il Loro Scopo:
-
HEPES: Mantiene la stabilità del pH.
-
Saccarosio / Mannitolo: Stabilizzatori osmotici per prevenire la rottura degli organelli.
-
EDTA / EGTA: Chelano i cationi bivalenti per inibire le metalloproteasi e prevenire la degradazione.
-
Inibitori delle proteasi e delle fosfatasi: Proteggono le proteine dalla degradazione enzimatica.
5. Note Critiche sulla Frazionamento del Campione
a. Attenzione alla perdita di proteine durante la centrifugazione:
-
Verificare se la proteina target è trattenuta nel pellet (detriti).
-
Esempio: Fino al 50% della miosina e circa il 66% di CSQ2 possono andare persi nei detriti se non controllati.
b. Verificare sempre se la proteina di interesse è:
-
Solubile (citoplasmatica)
-
Insolubile (legata al citoscheletro)
-
Associata a membrana (organellare)
Separazione Elettroforetica
Dopo l’estrazione e la quantificazione proteica, la fase successiva cruciale nell’analisi proteomica è la separazione elettroforetica. L’elettroforesi su gel rimane una tecnica fondamentale per migliorare selettività e sensibilità nella ricerca proteomica, consentendo la risoluzione di miscele proteiche complesse in base alle proprietà fisico-chimiche.

Vari matrici di gel—tra cui agarosio, amido e poliacrilammide—sono state sviluppate per applicazioni elettroforetiche. Tra queste, la poliacrilammide è la matrice di elezione per l’analisi proteica grazie alla sua stabilità meccanica, inerzia chimica e alla possibilità di regolare precisamente la polimerizzazione con bisacrilammide. Un rapporto acrilammide:bisacrilammide comunemente impiegato di 37.5:1 consente di ottenere gel robusti con dimensioni di poro riproducibili, adatti per una frazionamento proteico ad alta risoluzione.
Due forme principali di elettroforesi su gel di poliacrilammide (PAGE) sono ampiamente utilizzate: PAGE nativa e SDS-PAGE denaturante.
-
PAGE nativa conserva la struttura terziaria e quaternaria delle proteine, consentendo la separazione basata su una combinazione di carica netta, dimensione idrodinamica e conformazione. In sistemi tampone alcalini, la maggior parte delle proteine acquisisce una carica netta negativa, permettendo la migrazione verso l’anodo senza denaturazione. Un vantaggio principale della PAGE nativa è la conservazione dell’attività enzimatica e delle interazioni proteina–proteina native durante la separazione.
-
SDS-PAGE (elettroforesi su gel di poliacrilammide in presenza di SDS) è il metodo più comunemente utilizzato per separare proteine denaturate esclusivamente in base al peso molecolare. Il detergente anionico SDS si lega uniformemente lungo la catena polipeptidica, conferendo una carica negativa proporzionale alla lunghezza della proteina. In condizioni denaturanti (tipicamente con riscaldamento in 0,1% SDS), le proteine vengono linearizzate assumendo una forma allungata, permettendo la migrazione dipendente dalla dimensione attraverso la matrice di poliacrilammide. Le proteine più piccole attraversano il gel più rapidamente delle più grandi. Inoltre, la SDS-PAGE beneficia dei principi dell’isotacoforesi, concentrando i campioni proteici in bande strette e definite, migliorando così la risoluzione (fino a ≥ dieci volte in condizioni controllate).
L’attenzione nella preparazione del campione, composizione dei tamponi e caratteristiche del gel è essenziale per ottenere una separazione ottimale e riproducibile. I componenti dei tamponi di caricamento comunemente utilizzati e le rispettive funzioni sono riassunti nella Tabella 1.
Tabella 1. Tamponi di preparazione del campione comunemente usati. Localizzazione proteica - Tampone - Scopo
| Localizzazione proteica | Tampone | Scopo dei componenti (concentrazioni tipiche) |
| Cellula intera | Tampone non denaturante e non ionico: Tampone NP-40: 50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40 o Triton X-100 |
Tris-HCl (10–50 mM): mantiene il pH stabile NaCl (50–150 mM): riduce legami aspecifici, mantiene la forza ionica NP-40/Triton X-100 (0.1–1%): detergenti non ionici, solubilizzano proteine citosoliche e di membrana senza denaturarle, permeabilizzano le membrane e prevengono l’aggregazione proteica |
| Tampone denaturante: Tampone RIPA: 150 mM NaCl, 1% NP-40 o Triton X-100, 0.5% sodio deoxicolato, 0.1% SDS, 50 mM Tris, pH 8.0 |
Sodio deoxicolato (0.1–0.5%): detergente ionico, lisia le cellule e solubilizza componenti cellulari e di membrana SDS (0.1–1%): detergente ionico, provoca la distruzione delle membrane e linearizza le proteine legandosi ad esse |
|
| Citoplasmatiche (solubili) |
20 mM Tris-HCl, pH 7.5 o tampone NP-40 | Vedi sopra |
| Citoplasmatiche (legate al citoscheletro) |
Tampone PIPES-Triton: 10 mM PIPES, 50 mM KCl, 1% Triton X-100, 10 mM EGTA, 3 mM MgCl2, 2 M glicerolo |
PIPES (10 mM): tamponamento della soluzione KCl (50–150 mM): mantiene la forza ionica, riduce legami aspecifici EGTA (1–3 mM): inibizione delle metalloproteasi dipendenti da Ca2+ MgCl2 (1–5 mM): stabilizza alcuni complessi insieme a KCl Glicerolo (1–10%): stabilizza le proteine |
| Associata a membrana | Tampone RIPA | Vedi sopra |
| Mitocondriale | Tampone RIPA | Vedi sopra |
| Nucleare | Tampone RIPA | Vedi sopra; nota: NP-40 e Triton X-100 non riescono a lisare le membrane nucleari |
La percentuale di gel richiesta dipende dalla dimensione della proteina di interesse:
| Dimensione proteica | % Gel |
| 4–40 kDa | 20% |
| 12–45 kDa | 15% |
| 10–70 kDa | 12% |
| 15–100 kDa | 10% |
| 25–100 kDa | 8% |
| 60–210 kDa | 5% |
Possono essere utilizzati anche gel a gradiente.
| Dimensione proteica | % Gel |
| 5–200 kDa | Gradiente 4–12% |
| 4–200 kDa | Gradiente 4–20% |
| 3.5–110 kDa | Gradiente 10–20% |
Sistemi Tampone Continui vs Discontinui
I sistemi di separazione elettroforetica si classificano in continui e discontinui (detti anche multifasici o isotacoforetici), ciascuno con specifiche configurazioni tampone e dinamiche di separazione.
1. Sistemi tampone continui:
- Stessa composizione ionica e pH per campione, gel e serbatoi.
- Gel composto da una singola concentrazione di acrilammide.
- Le proteine si separano direttamente nel gel senza effetto stacking.
- Punto chiave: la risoluzione dipende fortemente da carichi campione piccoli e concentrati.
2. Sistemi tampone discontinui (multiphasici):
- Due tamponi con pH e forza ionica differenti:
- Gel di stacking: pH ~6.8, basso Tris–HCl (0.125 M), pori grandi.
- Gel di risoluzione: pH ~8.8, alto Tris–HCl (0.375 M), pori piccoli.
- Tampone di corsa (es. Tris-glicina) introdotto sopra il gel di stacking.
3. Meccanismo di separazione:
- Cl⁻ come ioni guida; glicinato come ioni di coda.
- Le proteine si concentrano in bande strette nel gel di stacking (“effetto stacking”).
- Nel gel di risoluzione, le proteine si separano in base al peso molecolare diventando gli ioni principali.
4. Vantaggi dei sistemi discontinui:
- Permettono il caricamento di volumi maggiori di campioni diluiti.
- Risoluzione superiore grazie allo stacking e alle diverse dimensioni dei pori del gel.
- Separazione più efficace basata sul peso molecolare.
Trasferimento delle Proteine
Dopo l'elettroforesi, le proteine vengono trasferite da un gel di poliacrilammide su una membrana. Questo passaggio è cruciale per immobilizzare le proteine in vista della successiva rilevazione e analisi (ad esempio, western blotting).
1. Membrane utilizzate per il trasferimento delle proteine
Esistono diverse membrane, ognuna con specifici vantaggi:
-
La nitrocellulosa è la membrana più comunemente utilizzata. Ha un'elevata affinità di legame per le proteine, può immobilizzare sia proteine che glicoproteine ed è compatibile con i metodi di rilevazione chemioluminescenti, cromogenici e fluorescenti. Tuttavia, è fragile perché derivata dalla cellulosa trattata con acido nitrico.
-
Il PVDF (polivinilidene difluoruro) offre una resistenza meccanica superiore rispetto alla nitrocellulosa, rendendolo ideale per operazioni di stripping e reprobing o per il sequenziamento delle proteine. Tuttavia, il PVDF può talvolta produrre una colorazione di fondo più elevata, richiedendo un'ottimizzazione supplementare.
2. Metodi di trasferimento delle proteine
Le proteine possono essere trasferite dal gel alla membrana mediante diversi metodi:
-
Trasferimento capillare basato sull'azione capillare passiva.
-
Trasferimento per diffusione che consente alle proteine di diffondere lentamente sulla membrana.
-
Elettrotrasferimento è il metodo più rapido e completo per il trasferimento.
Tra questi, l’elettrotrasferimento è il metodo preferito per la sua rapidità ed efficienza.
Tipi di elettrotrasferimento
Esistono tre tipi di elettrotrasferimento in base al metodo di utilizzo del tampone:
-
Elettrotrasferimento a secco utilizza componenti asciutti senza aggiunta di liquidi.
-
Elettrotrasferimento semi-secco impiega carte da filtro impregnate di tampone di trasferimento tra la membrana e il gel.
-
Elettrotrasferimento a umido prevede l'immersione dell'intero sandwich gel-membrana in un tampone di trasferimento, particolarmente raccomandato per il trasferimento di proteine di grandi dimensioni.
1. Principi dell'elettrotrasferimento
Durante l’elettrotrasferimento:
-
Viene applicato un campo elettrico perpendicolare al gel.
-
Le proteine, essendo cariche, migrano fuori dal gel verso la membrana.
-
Il gel deve essere orientato verso l’elettrodo negativo (catodo), mentre la membrana deve essere rivolta verso l’elettrodo positivo (anodo).
-
Per garantire un trasferimento corretto, gel e membrana devono essere ben pressati insieme, con carte da filtro e cuscinetti in fibra posizionati su entrambi i lati per evitare la formazione di bolle d’aria, che potrebbero causare un trasferimento non uniforme.
a. Tamponi di trasferimento
I tamponi di trasferimento aiutano a mantenere la struttura proteica e a migliorare il movimento durante il trasferimento. I tamponi più comuni includono:
-
Tampone Towbin (25 mM Tris, 192 mM glicina, 20% metanolo, pH 8.3) è ampiamente utilizzato per il trasferimento a umido.
-
Tampone Bjerrum Schafer-Nielsen (48 mM Tris, 39 mM glicina, pH 9.2, 20% metanolo) è preferito per il trasferimento semi-secco.
-
Tampone CAPS (60 mM Tris e 40 mM CAPS) è utilizzato soprattutto per le proteine con alto punto isoelettrico.
Il metanolo nel tampone aiuta a prevenire il rigonfiamento del gel e migliora il legame delle proteine con la membrana. Tuttavia, in alcuni casi, tamponi privi di metanolo possono comunque produrre segnali di rilevazione intensi.
Il ruolo dei componenti del tampone di trasferimento è descritto nella Tabella 2.
Tabella 2. Composizione dei tamponi di corsa e di trasferimento e ruolo di ciascun componente
| Componente potenziale | Ruolo |
| Tamponi di corsa | |
| Acetato | Buona separazione nella gamma di massa elevata (100–500 kDa) |
| Glicina | Fornisce ioni glicinato per formare l’estremità di inseguimento durante l’elettroforesi nel gel di stacking |
| MES | Necessario per mantenere un pH relativamente costante. Fornisce ioni MES per formare l’estremità di inseguimento durante l’elettroforesi. Il MES consente una separazione maggiore nella gamma di massa inferiore (<50 kDa). Il tampone MES-SDS è più rapido del tampone MOPS-SDS grazie al pKa più basso del MES rispetto al MOPS |
| MOPS | Necessario per mantenere un pH relativamente costante. Fornisce ioni MOPS per formare l’estremità di inseguimento durante l’elettroforesi. Il MOPS offre una separazione migliore nella gamma di massa media-alta. La differenza nella mobilità ionica tra MES e MOPS influisce sull’impilamento e quindi sulla gamma di separazione proteica tra i due tamponi |
| SDS | Aiuta a mantenere le proteine con carica netta negativa |
| Tricina | Necessaria per mantenere un pH relativamente costante. La tricina sostituisce la glicina nel tampone di corsa, fornendo ioni tricina per formare l’estremità di inseguimento durante l’elettroforesi. La tricina consente un impilamento e una risoluzione più efficaci delle proteine a basso peso molecolare. Ottima per separare proteine a basso peso molecolare (0.5–50 kDa) |
| Tris-HCl | Necessario per mantenere un pH relativamente costante. Fornisce ioni cloruro che formano l’estremità anteriore durante l’elettroforesi nel gel di stacking. Gli ioni idrogeno conducono elettricità |
| Tamponi di trasferimento | |
| Tris-HCl | Necessario per mantenere un pH relativamente costante |
| Glicina | In assenza di metanolo, aiuta a prevenire il rigonfiamento del gel |
| Metanolo | Previene il rigonfiamento del gel durante il trasferimento e migliora il legame delle proteine alla nitrocellulosa. Rimuove l’SDS dalle proteine permettendo un legame migliore alle membrane di nitrocellulosa. Alcuni laboratori non includono più il metanolo nei tamponi di trasferimento poiché i risultati senza metanolo non differivano significativamente da quelli con metanolo. Alcuni protocolli usano il 10% di etanolo invece del 20% di metanolo |
| SDS | L’SDS (fino allo 0,1%) nel tampone di trasferimento aumenta l’efficienza del trasferimento proteico, specialmente per le proteine di grandi dimensioni, ma può ridurre la quantità di proteina legata alla membrana. Nelle membrane di nitrocellulosa con pori più grandi (0,45 μm), le proteine denaturate di piccole dimensioni possono attraversare la membrana |
| CAPS | Necessario per mantenere un pH relativamente costante. Raccomandato per il trasferimento di proteine ad alto peso molecolare (>150 kDa) |
| CAPS: acido (cicloesilammino)-L-propansolfonico; MES: acido 2-(N-morfolino) etansolfonico; MOPS: acido 3-(N-morfolino) propansolfonico; SDS: Sodio Dodecil Solfato; Tris-HCl: Tris idrocloruro. |
|
b.Considerazioni Importanti
-
Proteine con peso molecolare molto basso (<10 kDa) possono essere trasferite in modo inefficiente.
-
Se il pH del tampone di trasferimento è inferiore al punto isoelettrico della proteina, le proteine possono migrare all’indietro.
-
Tamponi specializzati (come quelli a base di CAPS) sono spesso necessari per il trasferimento efficiente di proteine con un alto punto isoelettrico.
Fase di Blocco nel Western Blotting
Dopo il trasferimento riuscito delle proteine sulla membrana, il blocco rappresenta una fase cruciale per garantire la specificità e la chiarezza della successiva immunorilevazione.

1. Scopo e Importanza
-
Il blocco impedisce il legame aspecifico degli anticorpi alla membrana, riducendo così il rumore di fondo e prevenendo segnali falsi positivi.
-
Migliora l’accuratezza e l’affidabilità della rilevazione delle proteine durante il western blotting.
2. Procedura di Blocco
-
La membrana viene incubata per 1 ora a temperatura ambiente in una soluzione di blocco contenente:
-
Albumina Sierica Bovina (BSA) o latte scremato in polvere, diluiti in:
-
Tampone salino Tris con 0,1% di Tween 20 (TBST), oppure
-
Tampone salino fosfato con 0,1% di Tween 20 (PBST).
-
-
-
Le proteine presenti nella BSA e nel latte (principalmente caseina e proteine del siero) si legano ai siti non occupati della membrana, impedendo interazioni aspecifiche tra la membrana e gli anticorpi primari o secondari.
3. Caratteristiche della Membrana e Ruolo del Tween 20
-
Le membrane in nitrocellulosa e PVDF mostrano un’elevata affinità di legame per le proteine, prevalentemente tramite interazioni idrofobiche.
-
Sebbene le proteine della BSA e del latte possano associarsi debolmente alle proteine trasferite, l’aggiunta di Tween 20 al tampone di blocco riduce queste interazioni aspecifiche, garantendo un blocco più efficace.
Incubazione con Anticorpi e Rilevamento nel Western Blotting
1. Il Ruolo dell'Incubazione con Anticorpi nel Western Blotting
Dopo il blocco della membrana per prevenire legami aspecifici, il western blotting procede alla fase cruciale di incubazione con anticorpi. Inizialmente, la membrana viene incubata con un anticorpo primario che si lega specificamente alla proteina bersaglio. Dopo il lavaggio degli anticorpi non legati con tamponi come TBST o PBST, viene aggiunto un anticorpo secondario. Questo anticorpo riconosce l'anticorpo primario ed è tipicamente coniugato a un enzima come HRP (Perossidasi di Rafano) o Fosfatasi Alcalina (AP), permettendo il successivo rilevamento del segnale.
-
Anticorpi Primari: Fondamentali per il legame specifico e selettivo con le proteine bersaglio.
-
Anticorpi Secondari: Consentono il rilevamento e l'amplificazione del segnale.
Consiglio: È importante ottimizzare la concentrazione degli anticorpi per massimizzare la sensibilità e ridurre il rumore di fondo. Il nostro supporto tecnico è a disposizione per l'ottimizzazione del protocollo.
Le fasi di lavaggio sono fondamentali per rimuovere gli anticorpi non legati. Tuttavia, un lavaggio eccessivo (>20 minuti) può indebolire il segnale — quindi è essenziale un equilibrio nei tempi!
2. Metodi di Rilevamento del Segnale
Una volta che la proteina bersaglio è legata da anticorpi marcati, il segnale deve essere rilevato. Sono disponibili diversi metodi:
a. Chemiluminescenza (ECL)
La chemiluminescenza potenziata (ECL) è il metodo di rilevamento più diffuso. Utilizzando anticorpi secondari coniugati con HRP e substrati ECL, viene generata luce che può essere catturata su pellicola o mediante un sistema di acquisizione digitale.
-
Altamente sensibile
-
Ampio intervallo dinamico
-
Ideale per il rilevamento di proteine a bassa abbondanza
Prodotti Raccomandati:
Anticorpi Secondari coniugati HRP
Kit di Substrato ECL
Kit di Rilevamento ECL
b. Rilevamento Fluorescente
Gli anticorpi secondari coniugati a fluorocromi permettono la rilevazione multiplex — ovvero la visualizzazione simultanea di più proteine mediante fluorofori differenti.
-
Adatto per la rilevazione simultanea di più bersagli
-
Ottimo intervallo dinamico e quantificazione precisa
-
Ideale con fluorofori nell’intervallo infrarosso
Prodotti Raccomandati:
Anticorpi Secondari (Visibili e Infrarossi)
Sistemi di Imaging Fluorescente
Anticorpi Fluorescenti
c. Rilevamento Cromogenico
I metodi cromogenici utilizzano reazioni enzimatiche per generare un precipitato colorato visibile a occhio nudo direttamente sulla membrana.
- Ideale quando l’equipaggiamento per l’imaging è limitato.
-
Visualizzazione semplice
-
Non necessita di apparecchiature specializzate
-
Ideale per laboratori didattici e ricerche di base
Prodotti Raccomandati:
Kit di Substrati Cromogenici (es. BCIP/NBT)
d. Rilevamento Radioattivo (Oggi Meno Comune)
Le sonde radioattive erano ampiamente utilizzate in passato per il western blotting, ma oggi sono in gran parte sostituite da metodi più sicuri e altrettanto sensibili come ECL e fluorescenza. La scelta del metodo di rilevamento è cruciale per il successo del Western blot e dipende da sensibilità, specificità e tipo di analisi desiderata:
-
Massima sensibilità? → Scegliere il rilevamento tramite chemiluminescenza potenziata (ECL).
L’ECL offre elevata sensibilità e ampio intervallo dinamico, ideale per proteine poco abbondanti. L’intensità del segnale può essere acquisita su pellicole a raggi X o sistemi digitali. -
Analisi multiplex (rilevamento simultaneo di più bersagli)? → Scegliere il rilevamento fluorescente.
Gli anticorpi secondari fluorescenti permettono il rilevamento simultaneo di più proteine senza necessità di rimuovere e riapplicare la membrana. I segnali sono stabili, quantitativi e ideali per studi comparativi. -
Visualizzazione semplice e rapida? → Scegliere il rilevamento cromogenico.
I sistemi cromogenici utilizzano reazioni enzima-substrato per produrre un precipitato colorato sulla membrana. Sebbene meno sensibili rispetto a ECL o fluorescenza, sono facili da usare e non richiedono strumentazione specifica.
Inoltre, la scelta degli anticorpi primari e secondari, così come dei substrati o reagenti di rilevamento, deve essere compatibile con il metodo di rilevamento selezionato. L’abbinamento corretto è essenziale per ottenere segnali forti e specifici, riducendo al minimo il rumore di fondo.