Neuronal signaling
Neuronal signaling, also referred to as synaptic signaling, is the specialized mode of intercellular communication that enables rapid information transfer within the nervous system. Unlike autocrine or paracrine signaling, which rely on diffusion over relatively longer distances and timescales, synaptic signaling is characterized by direct, point-to-point transmission across specialized junctions called synapses (Südhof, 2013). This highly efficient mechanism allows neurons to integrate, process, and propagate electrical and chemical signals with millisecond precision, forming the foundation for sensation, movement, cognition, and memory.
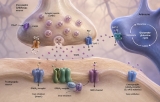
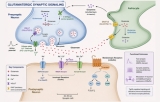
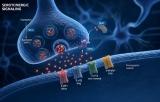
Two principal forms of synaptic signaling exist: chemical and electrical. Chemical synaptic transmission is the more prevalent form in the mammalian nervous system. Upon arrival of an action potential at the presynaptic terminal, voltage‑gated calcium channels open, triggering the fusion of synaptic vesicles with the presynaptic membrane and the release of neurotransmitters into the synaptic cleft (Katz & Miledi, 1967). Neurotransmitters—such as glutamate, γ‑aminobutyric acid (GABA), acetylcholine, or dopamine—then diffuse across the cleft, typically a distance of 20‑40 nm, and bind to ionotropic or metabotropic receptors on the postsynaptic membrane (Kandel et al., 2013). Ionotropic receptors directly open ion channels, generating fast excitatory or inhibitory postsynaptic potentials (EPSPs or IPSPs), while metabotropic receptors activate G‑protein‑dependent second messenger cascades that modulate synaptic strength over longer timescales (Conn & Pin, 1997). In contrast, electrical synaptic transmission is mediated by gap junctions formed by connexin proteins, which directly connect the cytoplasm of adjacent neurons, allowing passive diffusion of ions and small molecules (Bennett & Zukin, 2004). Electrical synapses are bidirectional and extremely fast, often serving to synchronize the activity of neuronal ensembles, such as in the inferior olive or during the generation of gamma oscillations.
A fundamental property of chemical synapses is synaptic plasticity, the activity‑dependent strengthening or weakening of synaptic transmission, which is widely considered the cellular correlate of learning and memory. Two best‑characterized forms are long‑term potentiation (LTP) and long‑term depression (LTD). In hippocampal CA1 pyramidal neurons, high‑frequency stimulation induces LTP through calcium influx via NMDA receptors, leading to activation of CaMKII and subsequent insertion of AMPA receptors into the postsynaptic membrane, thereby enhancing synaptic efficacy (Bliss & Collingridge, 1993; Lisman et al., 2012). Conversely, low‑frequency stimulation can induce LTD, which involves protein phosphatases (e.g., calcineurin) and internalization of AMPA receptors (Malenka & Bear, 2004). Beyond glutamate synapses, plasticity also occurs at inhibitory GABAergic synapses and at neuromodulatory synapses involving dopamine or serotonin, each governed by distinct molecular rules (Castillo et al., 2011).
The precise spatial organization of synaptic signaling is critical for its function. The postsynaptic density (PSD), an electron‑dense protein complex located directly opposite the presynaptic active zone, clusters receptors, adhesion molecules, and scaffolding proteins (e.g., PSD‑95) to ensure efficient signal reception and transduction (Sheng & Hoogenraad, 2007). Trans‑synaptic cell adhesion molecules, such as neurexins and neuroligins, bridge the pre‑ and postsynaptic compartments and are essential for synapse formation, specification, and maintenance (Südhof, 2008). Dysfunction of these molecular components is implicated in neurodevelopmental disorders, including autism spectrum disorder and schizophrenia (Bourgeron, 2015).
Quantitative aspects of synaptic signaling have been extensively modeled. The probability of neurotransmitter release (Pr) is a key parameter that varies across synapses and is modulated by presynaptic calcium dynamics, vesicle pool size, and the presence of presynaptic receptors for retrograde messengers (Dobrunz & Stevens, 1997). Classic quantal analysis demonstrates that neurotransmitter release occurs in discrete packets (quanta), each corresponding to the contents of a single synaptic vesicle (Del Castillo & Katz, 1954). Moreover, synapses exhibit short‑term plasticity—facilitation, depression, and augmentation—lasting milliseconds to seconds, which underlies adaptive processes such as adaptation, gain control, and temporal filtering (Zucker & Regehr, 2002).
Pathologically, disrupted synaptic signaling is central to numerous neurological and psychiatric disorders. In Alzheimer's disease, early synaptic dysfunction, particularly loss of glutamate receptors and impairment of LTP, precedes frank neurodegeneration (Selkoe, 2002). Mutations in genes encoding synaptic proteins, such as SHANK3 and SYNGAP1, are causative for intellectual disability and autism (Südhof, 2008). Additionally, abnormal dopamine signaling at synapses in the ventral tegmental area and nucleus accumbens is a core feature of addiction and Parkinson's disease (Beaulieu & Gainetdinov, 2011). Consequently, many therapeutic agents—including benzodiazepines (acting on GABA receptors), antidepressants (modulating monoamines), and anticonvulsants (targeting ion channels)—directly modulate synaptic signaling.
In summary, neuronal (synaptic) signaling represents a highly specialized, rapid, and plastic form of cell–cell communication that converts electrical impulses into chemical messages and back. Its molecular machinery—comprising voltage‑gated channels, synaptic vesicles, neurotransmitter receptors, and adhesion molecules—operates with exquisite spatial and temporal precision. Understanding the principles of synaptic transmission and plasticity remains a central goal of neuroscience, with profound implications for treating brain disorders and for the development of neuromorphic computing systems.
References
Beaulieu, J. M., & Gainetdinov, R. R. (2011). The physiology, signaling, and pharmacology of dopamine receptors. Pharmacological Reviews, 63(1), 182–217.
Bennett, M. V. L., & Zukin, R. S. (2004). Electrical coupling and neuronal synchronization in the mammalian brain. Neuron, 41(4), 495–511.
Bliss, T. V. P., & Collingridge, G. L. (1993). A synaptic model of memory: Long‑term potentiation in the hippocampus. Nature, 361(6407), 31–39.
Bourgeron, T. (2015). From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nature Reviews Neuroscience, 16(9), 551–563.
Castillo, P. E., Chiu, C. Q., & Carroll, R. C. (2011). Long‑term plasticity at inhibitory synapses. Current Opinion in Neurobiology, 21(2), 328–338.
Conn, P. J., & Pin, J. P. (1997). Pharmacology and functions of metabotropic glutamate receptors. Annual Review of Pharmacology and Toxicology, 37, 205–237.
Del Castillo, J., & Katz, B. (1954). Quantal components of the end‑plate potential. The Journal of Physiology, 124(3), 560–573.
Dobrunz, L. E., & Stevens, C. F. (1997). Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron, 18(6), 995–1008.
Kandel, E. R., Schwartz, J. H., Jessell, T. M., Siegelbaum, S. A., & Hudspeth, A. J. (2013). Principles of Neural Science (5th ed.). McGraw‑Hill.
Katz, B., & Miledi, R. (1967). The timing of calcium action during neuromuscular transmission. The Journal of Physiology, 189(3), 535–544.
Lisman, J., Yasuda, R., & Raghavachari, S. (2012). Mechanisms of CaMKII action in long‑term potentiation. Nature Reviews Neuroscience, 13(3), 169–182.
Malenka, R. C., & Bear, M. F. (2004). LTP and LTD: An embarrassment of riches. Neuron, 44(1), 5–21.
Selkoe, D. J. (2002). Alzheimer's disease is a synaptic failure. Science, 298(5594), 789–791.
Sheng, M., & Hoogenraad, C. C. (2007). The postsynaptic architecture of excitatory synapses: A more quantitative view. Annual Review of Biochemistry, 76, 823–847.
Südhof, T. C. (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature, 455(7215), 903–911.
Südhof, T. C. (2013). Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron, 80(3), 675–690.
Zucker, R. S., & Regehr, W. G. (2002). Short‑term synaptic plasticity. Annual Review of Physiology, 64, 355–405.