
Protocolo de Reacción en Cadena de la Polimerasa (PCR)
Introducción
La Reacción en Cadena de la Polimerasa (PCR) es una técnica fundamental en biología molecular que permite la amplificación de secuencias de ADN específicas a partir de cantidades mínimas de material inicial. Desde su invención por Kary Mullis en 1983, la PCR ha revolucionado la investigación genética, el diagnóstico, la ciencia forense y la biotecnología. Este protocolo proporciona una guía completa para realizar una PCR estándar, incluyendo pasos detallados, especificaciones de reactivos, resolución de problemas y estrategias de optimización.
Principio de la PCR
La PCR es un proceso enzimático que amplifica un segmento de ADN específico mediante ciclos repetidos de desnaturalización, hibridación y extensión. La reacción depende de una polimerasa de ADN termoestable, típicamenteFACILmente Taq, que soporta las altas temperaturas necesarias para desnaturalizar el ADN de doble cadena (Figura 1).
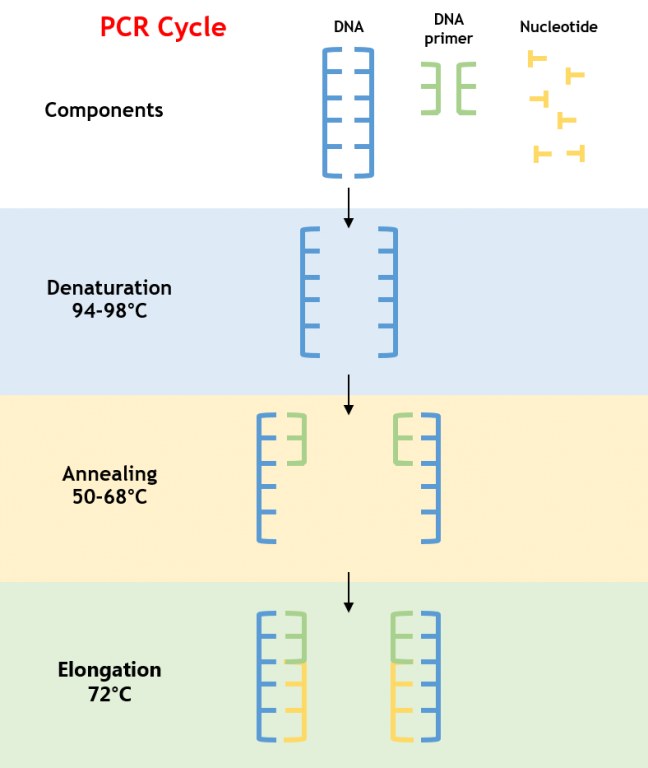
Figura 1: Proceso de Amplificación de la PCR
- Desnaturalización: El ADN se calienta a 94–98°C, rompiendo los enlaces de hidrógeno para producir ADN de cadena simple.
- Hibridación: La temperatura se reduce a 50–68°C, permitiendo que los cebadores se unan a las secuencias complementarias que flanquean el ADN objetivo.
- Extensión: La temperatura se eleva a 72°C, óptima para que la polimerasa Taq sintetice nuevas cadenas de ADN añadiendo desoxinucleótidos trifosfatos (dNTPs).
Materiales y Equipos
Antes de comenzar, asegúrese de que todos los reactivos y equipos estén disponibles y almacenados adecuadamente. Utilice reactivos de grado de biología molecular para evitar contaminación o inhibición.
Reactivos
- Polimerasa de ADN Taq: Almacenada a -20°C.
- Buffer de PCR 10X: Normalmente incluye 100 mM Tris-HCl, 500 mM KCl y 15 mM MgCl₂ (pH 8.3–8.8). Algunos buffers no incluyen MgCl₂, lo que requiere su adición por separado. Almacenar a -20°C o 4°C según el fabricante.
- Cloruro de Magnesio: Solución de MgCl₂ 25 mM (si no está incluido en el buffer de PCR). Almacenar a -20°C o 4°C.
- dNTPs: Desoxinucleótidos trifosfatos (dATP, dTTP, dCTP, dGTP; 10 mM cada uno, combinados o separados). Almacenar a -20°C.
- Cebadores: Cebadores forward y reverse (100 µM en buffer TE, diluidos a 10 µM en solución de trabajo).
- ADN Molde: ADN genómico (10–100 ng), ADN plasmídico (0.1–1 ng), o cDNA (1–10 ng), dependiendo de la aplicación. Almacenar a -20°C.
- Agua Libre de Nucleasas: Agua estéril de grado de biología molecular. Almacenar a temperatura ambiente.
- Mezcla Maestra
- Tinte de Gel de ADN SYBR® Safe: Para visualización de gel de agarosa (alternativa: bromuro de etidio, pero use con precaución debido a su toxicidad). Almacenar a 4°C.
- Tinte de Carga 6X: Para electroforesis de gel (por ejemplo, contiene azul de bromofenol y cianuro de xileno). Almacenar a 4°C.
- Escalera de ADN: Escalera de 100 bp o 1 kb, dependiendo del tamaño esperado del amplicón. Almacenar a -20°C.
Equipos
- Ciclador Térmico: Máquina de PCR programable.
- Tubos de PCR: Tubos de PCR de pared fina de 0.2 mL o placas de 96 pozos, compatibles con el ciclador térmico.
- Pipetas y Puntas: Pipetas calibradas (P2, P10, P20, P200, P1000) con puntas de filtro para evitar contaminación.
- Microcentrífuga: Para breves centrifugaciones para recolectar componentes de la reacción.
- Sistema de Electroforesis de Gel de Agarosa: Incluye bandeja de colado de gel, peine, fuente de alimentación y transiluminador UV (o luz azul para SYBR Safe).
- Mezclador de Vórtice: Para mezclar reactivos.
- Cubo de Hielo: Para mantener los reactivos fríos durante la preparación.
- Espectrofotómetro: Para cuantificar la concentración de ADN.
Protocolo de PCR
Paso 1: Diseño y Preparación de Cebadores
Los cebadores son oligonucleótidos de ADN de cadena simple (18–25 bases) que flanquean la secuencia objetivo y sirven como puntos de partida para la síntesis de ADN. El diseño adecuado de los cebadores es crítico para una amplificación específica y eficiente.
Directrices para el Diseño de Cebadores
Utilice herramientas de software para diseñar cebadores. Siga estos criterios:
- Longitud: 18–25 bases. Los cebadores más cortos (<18) pueden carecer de especificidad; los más largos (>25) pueden formar estructuras secundarias.
- Temperatura de Fusión (Tm): 55–65°C, con cebadores forward y reverse dentro de 2°C entre sí. Calcule Tm usando la fórmula para una estimación básica, o utilice software para mayor precisión (considera la concentración de sal y cebadores).
Tm = 4 (G+C) + 2 (A+T)
- Contenido de GC: 40–60%. Evite extremos (<30% o >70%) para prevenir una hibridación deficiente o estructuras secundarias.
- Extremo 3': Termine con una G o C (unión de bases más fuerte) y evite secuencias de tres o más bases idénticas (por ejemplo, GGG) para reducir errores de hibridación.
- Especificidad: Verifique la especificidad usando NCBI BLAST (blast.ncbi.nlm.nih.gov) para asegurar que los cebadores se unan solo a la secuencia objetivo. Evite homología con regiones no objetivo.
- Estructuras Secundarias: Evite horquillas, dímeros propios y dímeros heterogéneos. El Delta G para dímeros debe ser >-5 kcal/mol.
- Tamaño del Amplicón: Diseñe para amplicones de 100–1000 bp para PCR estándar. Los amplicones más largos (hasta 5 kb) son posibles pero requieren optimización.
- Evite Repeticiones: No coloque cebadores en regiones con secuencias repetitivas o de baja complejidad (por ejemplo, ATATAT).
Síntesis y Almacenamiento de Cebadores
- Pedido: Solicite cebadores purificados por HPLC. La purificación por HPLC asegura alta pureza, reduciendo la amplificación no específica.
- Resuspensión: Resuspenda los cebadores liofilizados en buffer TE (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) a una concentración de 100 µM.
- Solución de Trabajo: Diluya el stock a 10 µM en agua libre de nucleasas. Almacene a -20°C.
- Almacenamiento: Almacene los stocks de 100 µM a -20°C por hasta 1 año; las soluciones de trabajo de 10 µM son estables a -20°C por 6 meses o a 4°C por 1 mes. Evite ciclos de congelación-descongelación repetidos (>10) para prevenir degradación.
Control de Calidad de los Cebadores
- Cuantificación: Verifique la concentración de los cebadores usando un espectrofotómetro (A260). La absorbancia esperada para 100 µM es ~0.3–0.5, dependiendo de la secuencia.
- Integridad: Opcional—corra los cebadores en un gel de agarosa al 2% para confirmar que no hay degradación (debería aparecer como una banda única y nítida).
- Prueba de Amplificación: Realice una PCR de prueba con una plantilla conocida para confirmar la funcionalidad de los cebadores antes de experimentos a gran escala.
Paso 2: Configuración de la Reacción
La mezcla de reacción de PCR contiene todos los componentes necesarios para la amplificación de ADN. El protocolo proporcionado está escalado a un volumen de reacción de 50 µL, con una mezcla maestra para 11 reacciones (10 muestras + 1 control sin plantilla) para compensar las pérdidas por pipeteo.
Componentes de la Reacción
La tabla a continuación enumera los reactivos y volúmenes para una reacción de 50 µL y la mezcla maestra para 11 reacciones.
| Reactivo | Volumen por Reacción (µL) | Total para 11 Reacciones (µL) |
| Agua Libre de Nucleasas | 36.5 | 401.5 |
| Buffer de PCR 10X | 5.0 | 55.0 |
| dNTPs (10 mM cada uno) | 1.0 | 11.0 |
| MgCl₂ (25 mM) | 3.0 | 33.0 |
| Cebador Forward (10 µM) | 1.0 | 11.0 |
| Cebador Reverse (10 µM) | 1.0 | 11.0 |
| Polimerasa Taq (5 U/µL) | 0.5 | 5.5 |
| ADN Molde (variable) | 3.0 | Añadir por separado |
Paso 3: Ciclo Térmico
El ciclador térmico somete la reacción a cambios de temperatura repetidos para facilitar la desnaturalización, hibridación y extensión. Las condiciones de ciclo proporcionadas están optimizadas para PCR estándar con polimerasa Taq y amplicones de 100–1000 bp.
|
|
|
|
|
NeoCycler Trio - Ciclador Térmico Multibloque (Cat# NB-12-3024) NeoCycler Duo - Ciclador Térmico Multibloque (Cat# NB-12-3025) |
NeoCycler 300 - Ciclador Térmico de Gradiente Rápido (con módulo 9677 / 96) (Cat# NB-12-3001) NeoCycler 200 - Ciclador Térmico de Gradiente (Cat# NB-12-3002) NeoCycler 100 - Ciclador Térmico sin Gradiente (con módulo 9677 / 96) (Cat# NB-12-3003) |
|
|
|
|
|
|
NeoCycler 600 - Ciclador Térmico de Súper Gradiente (Cat# NB-12-3026) |
NeoCycler Mini (Cat# NB-12-3029-2) |




Programa de Ciclo Térmico
Programe el ciclador térmico con los siguientes pasos:
- Desnaturalización Inicial: 95°C durante 2 minutos
- Desnaturaliza el ADN molde de doble cadena y activa la polimerasa Taq (las versiones de arranque en caliente pueden requerir más tiempo, por ejemplo, 5–10 minutos).
- Ciclos (30–35 ciclos):
- Desnaturalización: 95°C durante 30 segundos
- Separa las cadenas de ADN. Tiempos más cortos (15–20 segundos) pueden ser suficientes para plantillas pequeñas (por ejemplo, plásmidos).
- Hibridación: 55–65°C durante 30 segundos
- Los cebadores se unen a las secuencias complementarias. Configure 2–5°C por debajo del Tm del cebador más bajo (por ejemplo, si Tm es 60°C, use 55–58°C). Optimice si es necesario (ver Sección 8).
- Extensión: 72°C durante 1 minuto por kb
- La polimerasa Taq sintetiza nuevo ADN. Para un amplicón de 500 bp, use 30–60 segundos; para 1 kb, use 60 segundos. Mínimo 30 segundos para <500 bp.
- Desnaturalización: 95°C durante 30 segundos
- Extensión Final: 72°C durante 5 minutos
- Completa extensiones parciales y agrega salientes 3' A (útil para clonación TA).
- Mantenimiento: 4°C indefinidamente
- Almacena las reacciones hasta su recuperación. Evite el almacenamiento prolongado (>24 horas) para prevenir la degradación.
Número de Ciclos
- Estándar: 30 ciclos para cantidades moderadas de plantilla (10–50 ng de ADN genómico).
- Plantilla Baja: 35 ciclos para cantidades bajas de plantilla (<1 ng) o cDNA.
- Plantilla Alta: 25–28 ciclos para cantidades altas de plantilla (>100 ng) para reducir productos no específicos.
- Ciclos Excesivos: >35 ciclos pueden aumentar la amplificación no específica o dímeros de cebadores.
Configuraciones del Ciclador Térmico
- Temperatura de la Tapa: Configure a 105°C para evitar la condensación.
- Tasa de Rampa: Use configuraciones predeterminadas (2–5°C/segundo). Las rampas más rápidas pueden requerir ajustes para máquinas más antiguas.
- Volumen: Configure a 50 µL en el programa del ciclador para un calentamiento preciso.
Variaciones
- Amplicones Cortos (<200 bp): Reduzca el tiempo de extensión a 15–30 segundos.
- Amplicones Largos (1–5 kb): Aumente el tiempo de extensión (1–2 minutos/kb) y considere polimerasas de alta fidelidad.
- Plantillas Ricas en GC: Use temperaturas de desnaturalización más altas (98°C) o aditivos como DMSO (5–10%).
Paso 4: Análisis de Productos de PCR
Después de la PCR, analice los productos para confirmar el éxito de la amplificación y el tamaño del amplicón. La electroforesis de gel de agarosa es el método estándar.
Preparación de Gel de Agarosa
- Concentración del Gel:
- 1% de agarosa para amplicones de 500–5000 bp.
- 1.5–2% de agarosa para amplicones de 100–500 bp.
- Preparar Gel:
- Pese la agarosa.
- Disuelva en buffer TAE 1X (40 mM Tris, 20 mM ácido acético, 1 mM EDTA) calentando en microondas hasta que esté claro.
- Enfríe a ~50°C, añada SYBR Safe (1 µL por 10 mL de gel, según el fabricante), y vierta en una bandeja de colado con un peine.
- Deje solidificar durante 20–30 minutos.
- Configuración: Coloque el gel en un tanque de electroforesis lleno de buffer TAE 1X.
Preparación de Muestras
- Mezclar Muestra: Combine 10 µL de producto de PCR con 2 µL de tinte de carga 6X (final 1X).
- Cargar Escalera: Cargue 5–10 µL de escalera de ADN (100 bp o 1 kb, dependiendo del tamaño del amplicón) en un carril.
- Cargar Muestras: Cargue 12 µL de cada mezcla de producto de PCR + tinte en pozos separados. Incluya el Control Sin Plantilla (NTC) para verificar la contaminación.
Electroforesis
- Correr Gel: Corra a 80–120 V durante 30–60 minutos, hasta que el tinte de carga (azul de bromofenol) migre ~2/3 de la longitud del gel.
- Visualizar: Use un transiluminador UV (o luz azul para SYBR Safe) para visualizar las bandas. Fotografíe el gel para documentarlo.
Interpretación
- Banda Esperada: Compare el tamaño de la banda con la escalera de ADN. Debe coincidir con el tamaño del amplicón predicho (basado en el diseño de cebadores).
- NTC: No deben aparecer bandas en el carril del NTC. Las bandas indican contaminación o dímeros de cebadores.
- Difuminado: Indica amplificación no específica, plantilla degradada o exceso de ADN.
- Sin Bandas: Sugiere una amplificación fallida.
Post-Análisis
- Almacenamiento: Almacene los productos de PCR a -20°C durante semanas o a 4°C durante 1–2 días.
- Purificación: Para aplicaciones posteriores (por ejemplo, secuenciación, clonación), purifique los productos usando un kit de limpieza de PCR o un kit de extracción de gel.
- Cuantificación: Mida la concentración del producto usando un Nanodrop o fluorómetro si es necesario.
Control de Calidad y Validación
Para asegurar resultados confiables, incorpore controles de calidad en cada paso.
Calidad de la Plantilla
- Pureza: Una relación A260/A280 de 1.8–2.0 indica ADN puro. Relaciones más bajas sugieren contaminación por proteínas o ARN.
- Integridad: Corra el ADN de la plantilla en un gel de agarosa al 0.8% para confirmar que no hay degradación (el ADN genómico debe aparecer como una banda de alto peso molecular).
- Concentración: Use 10–100 ng de ADN genómico o 0.1–1 ng de ADN plasmídico por reacción de 50 µL.
Calidad de los Reactivos
- dNTPs: Verifique la degradación (almacene a -20°C, evite >10 ciclos de congelación-descongelación).
- Polimerasa Taq: Verifique la actividad con una plantilla-cebador conocida.
- Cebadores: Confirme la concentración y la integridad.
Controles
- NTC: Detecta contaminación o dímeros de cebadores.
- Control Positivo: Use una plantilla-cebador conocida para confirmar las condiciones de la reacción.
- Control Negativo (opcional): Omita la polimerasa para verificar la amplificación no específica.
Análisis de Gel
- Escalera: Asegúrese de que la escalera se resuelva claramente para confirmar la calidad del gel.
- Intensidad de la Banda: Bandas fuertes y únicas indican una amplificación exitosa. Bandas débiles o múltiples sugieren que se necesita optimización.
Resolución de Problemas
La PCR puede requerir optimización para lograr una amplificación específica y de alto rendimiento. A continuación, se presentan problemas comunes y soluciones.
Sin Amplificación
- Causa: Baja concentración de plantilla, plantilla degradada, cebadores incorrectos o temperatura de hibridación subóptima.
- Soluciones:
- Aumente la plantilla (hasta 200 ng) o los ciclos (hasta 35).
- Verifique la integridad de la plantilla en un gel.
- Confirme las secuencias de los cebadores y Tm; rediseñe si es necesario.
- Realice una PCR de gradiente (50–65°C de hibridación) para encontrar la temperatura óptima.
- Verifique la actividad de la polimerasa Taq con un control positivo.
Bandas No Específicas
- Causa: Temperatura de hibridación baja, exceso de MgCl₂, exceso de cebadores o unión no específica de cebadores.
- Soluciones:
- Aumente la temperatura de hibridación en incrementos de 1–2°C.
- Reduzca el MgCl₂ a 1–1.2 mM.
- Disminuya la concentración de cebadores a 0.1–0.15 µM.
- Rediseñe los cebadores para mayor especificidad (use BLAST).
- Reduzca el número de ciclos a 25–28.
Difuminado
- Causa: Exceso de plantilla, plantilla degradada o exceso de ciclos.
- Soluciones:
- Reduzca la plantilla a 10–50 ng.
- Verifique la integridad de la plantilla.
- Disminuya los ciclos a 25–30.
- Use dNTPs y polimerasa frescos.
Dímeros de Cebadores
- Causa: Los cebadores forman dímeros propios o heterogéneos, visibles como bandas de bajo peso molecular (<100 bp) en el NTC.
- Soluciones:
- Rediseñe los cebadores para minimizar la formación de dímeros.
- Reduzca la concentración de cebadores a 0.1 µM.
- Aumente la temperatura de hibridación.
- Use polimerasa Taq de arranque en caliente para prevenir la hibridación no específica durante la configuración.
Estrategias de Optimización
- PCR de Gradiente: Pruebe un rango de temperaturas de hibridación (por ejemplo, 50–65°C) en una sola corrida si el ciclador soporta la función de gradiente.
- Titulación de MgCl₂: Pruebe 1–4 mM de MgCl₂ en incrementos de 0.5 mM.
- Aditivos: Para plantillas ricas en GC, añada 5–10% de DMSO o betaína (1–2 M) a la reacción.
- PCR de Arranque en Caliente: Use Taq de arranque en caliente (por ejemplo, Platinum Taq) para reducir la amplificación no específica durante la configuración.
- PCR de Touchdown: Comience la hibridación a 5–10°C por encima del Tm, disminuyendo 0.5°C por ciclo durante 10 ciclos, luego continúe a la temperatura más baja. Mejora la especificidad.
Conclusión
Este protocolo integral de PCR proporciona un marco robusto para amplificar ADN con alta especificidad y rendimiento. Siguiendo los pasos detallados para el diseño de cebadores, la configuración de la reacción, el ciclo térmico y el análisis de productos, los usuarios pueden lograr resultados confiables para una amplia gama de aplicaciones. Los consejos de optimización y resolución de problemas aseguran el éxito incluso con plantillas o cebadores desafiantes. Para aplicaciones avanzadas, adapte el protocolo según se describe, y consulte las directrices del fabricante para reactivos o equipos especializados.