Western-Blot-Protokoll

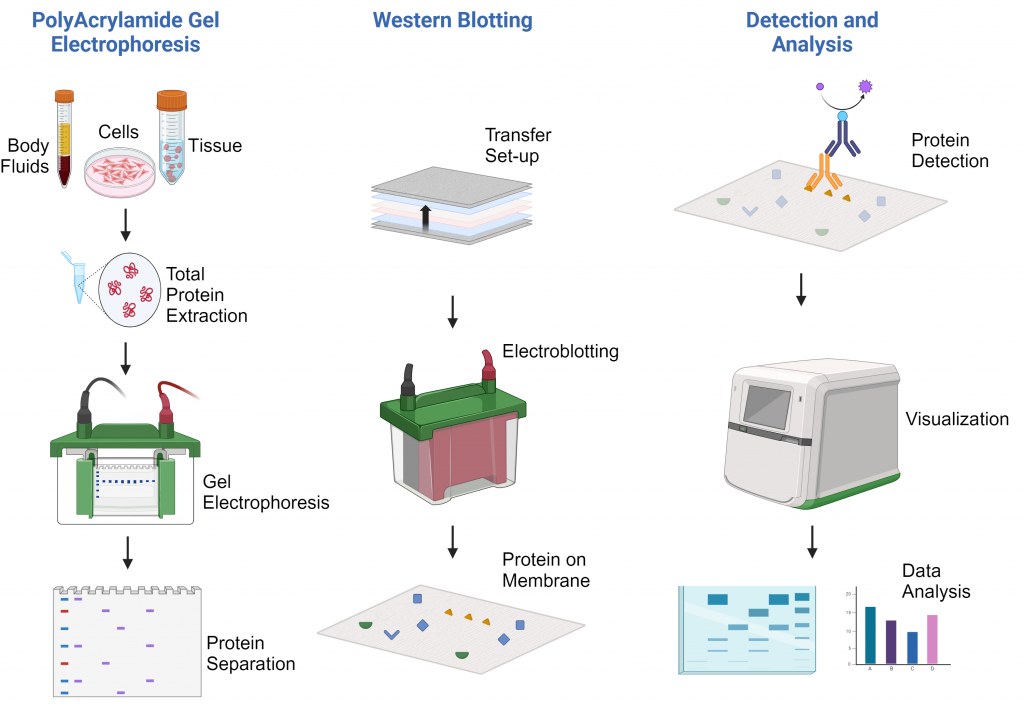
Prinzip der Technik
Das Prinzip des Western Blotting umfasst mehrere wichtige Schritte:
-
Proteintrennung: Proteine werden mittels SDS-PAGE nach ihrer Größe getrennt.
-
Transfer: Die getrennten Proteine werden auf eine feste Membran (Nitrocellulose oder PVDF) übertragen.
-
Blockierung: Die Membranoberflächen werden blockiert, um eine unspezifische Bindung zu verhindern.
-
Nachweis: Spezifische Primärantikörper erkennen die Zielproteine, gefolgt von markierten Sekundärantikörpern.
-
Visualisierung: Protein-Antikörper-Komplexe werden durch chemilumineszente, fluoreszierende oder kolorimetrische Reaktionen sichtbar gemacht.
Der Erfolg dieser Technik hängt in hohem Maße von der Spezifität der Antikörper und der Unversehrtheit der Probenvorbereitung ab.
Der erste wichtige Schritt, der beim Western Blotting nicht übersehen werden darf, ist die Probenvorbereitung. Um reproduzierbare Ergebnisse zu erhalten, sollte eine effiziente Proteinextraktion und -reinigung mit einer geeigneten Homogenisierungsmethode durchgeführt werden, die den intrazellulären Inhalt der Zelle durch Aufbrechen der Zellmembranen effizient freisetzen kann.
Proteinprobenisolierung aus verschiedenen Arten von Proben
Die Proteinextraktion und -isolierung sind wichtige erste Schritte in einer Vielzahl von biochemischen und molekularbiologischen Verfahren, darunter Western Blotting, Massenspektrometrie, Enzymassays und Immunpräzipitation. Der Erfolg der nachgelagerten Anwendungen hängt in hohem Maße von der Qualität, Integrität und Repräsentativität der isolierten Proteinproben ab.
Biologische Proben wie kultivierte Zellen, Gewebe, Bakterien, Hefe und Pflanzenmaterial stellen jeweils besondere Herausforderungen für die Proteinextraktion dar. Daher ist die Wahl einer geeigneten Isolierungsstrategie entscheidend, um die Ausbeute zu maximieren, die Proteinfunktionalität zu erhalten und den Abbau oder die Veränderung zu minimieren.
1. Homogenisierung
-
Wählen Sie eine Homogenisierungsmethode entsprechend der Art der Probe:
-
Ultraschallbehandlung (für empfindliche Proben)
-
French Press (für Bakterien, Hefe)
-
Glasperlenmühlen (für zähe Zellen, Pilze)
-
Dounce/Potter-Elvehjem-Homogenisator (für weiches Gewebe)
-
Manuelles Zerkleinern mit Mörser und Stößel (für gefrorenes Gewebe)
-
-
Ziel: Effiziente Zelllyse ohne Proteabbau.
2. Handhabung der Proben nach der Behandlung
-
Nach Exposition gegenüber Xenobiotika oder genetischer Manipulation (z. B. siRNA-Knockdown):
-
Proben sofort in flüssigem Stickstoff einfrieren.
-
Lysat sofort, um eine endogene Proteaseaktivität zu verhindern.
-
- Proben während der Verarbeitung stets kühl halten.
3. Frost-Tau-Zyklus-Management
- Vermeiden Sie mehrfache Einfrier- und Auftauzyklen, um die Proteinintegrität zu schützen.
-
-
Proben aliquotieren, um unnötiges Auftauen zu vermeiden.
-
4. Auswahl des Lysepuffers
a. Puffer an die Position des Zielproteins anpassen:
-
Cytosolic proteins → Mild lysis buffer (e.g., NP-40, Triton X-100)
-
Membrangebundene Proteine → Stärkere Puffer (z. B. RIPA)
-
Nukleare/mitochondriale Proteine → Spezialisierte Puffer (z. B.Hypotonischer Puffer, Drosophila-Homogenisierungspuffer)
b. Puffer an die Empfindlichkeit des Antikörpers anpassen:
- Native Proteinnachweis → Mildes/ nichtionisches Reinigungsmittel oder kein Reinigungsmittel
-
Nachweis von denaturierten Proteinen → Starke/denaturierende Bedingungen
-
Fügen Sie Protease- und Phosphatase-Inhibitoren allen Lysepuffern hinzu.
c. Einige häufige Inhaltsstoffe und warum sie verwendet werden:
-
HEPES: Erhält die pH-Stabilität.
-
Saccharose / Mannitol: Osmotische Stabilisatoren, um das Platzen von Organellen zu verhindern.
-
EDTA / EGTA: Chelatisiert zweiwertige Kationen, um Metalloproteasen zu hemmen und den Abbau zu verhindern.
-
Protease- und Phosphatase-Inhibitoren: Schützen Proteine vor enzymatischem Abbau.
5. Kritische Anmerkungen zur Probenfraktionierung
a. Achten Sie auf Proteinverlust während der Zentrifugation:
-
Testen Sie, ob das Zielprotein im Pellet (Debris) zurückbleibt.
- Beispiel: Bis zu 50 % des Myosins und ~66 % des CSQ2 gingen in den Rückständen verloren, wenn dies nicht überprüft wurde.
b. Überprüfen Sie immer, ob das gesuchte Protein:
- Löslich (zytoplasmatisch)
- Unlöslich (Zytoskelett)
- Membrangebunden (organellär)
Elektrophoretische Trennung
Nach der Proteinextraktion und -quantifizierung ist die nächste entscheidende Phase der Proteomanalyse die elektrophoretische Trennung. Die Gelelektrophorese ist nach wie vor eine grundlegende Technik zur Verbesserung der Selektivität und Empfindlichkeit in der Proteomforschung, da sie die Auflösung komplexer Proteinmischungen auf der Grundlage physikalisch-chemischer Eigenschaften ermöglicht.

Für elektrophoretische Anwendungen wurden verschiedene Gelmatrizen entwickelt, darunter Agarose, Stärke und Polyacrylamid. Unter diesen ist Polyacrylamid aufgrund seiner mechanischen Stabilität, chemischen Trägheit und der Möglichkeit, die Polymerisation mit Bisacrylamid präzise zu regulieren, die Matrix der Wahl für die Proteinanalyse. Ein häufig verwendetes Acrylamid:Bisacrylamid-Verhältnis von 37,5:1 ermöglicht die Herstellung robuster Gele mit reproduzierbaren Porengrößen, die für die hochauflösende Proteinfraktionierung geeignet sind.
Zwei Hauptformen der Polyacrylamid-Gelelektrophorese (PAGE) sind weit verbreitet: native PAGE und denaturierende SDS-PAGE.
-
Native PAGE erhält die Tertiär- und Quartärstrukturen von Proteinen und ermöglicht deren Trennung anhand einer Kombination aus Nettoladung, hydrodynamischer Größe und Konformation. In alkalischen Puffersystemen erhalten die meisten Proteine eine negative Nettoladung, wodurch sie ohne Denaturierung zur Anode wandern können. Ein wesentlicher Vorteil der nativen PAGE ist die Erhaltung der enzymatischen Aktivität und der nativen Protein-Protein-Wechselwirkungen während der Trennung.
-
SDS-PAGE (Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis) ist die am häufigsten verwendete Methode zur Trennung denaturierter Proteine ausschließlich anhand ihres Molekulargewichts. Das anionische Detergens SDS bindet sich gleichmäßig entlang der Polypeptidkette und verleiht ihr eine konstante negative Ladung, die proportional zur Proteingröße ist. Unter denaturierenden Bedingungen (in der Regel durch Erhitzen in 0,1 % SDS) werden Proteine linearisiert und nehmen eine gestreckte, stabförmige Form an, wodurch sie je nach Größe durch die Polyacrylamidmatrix wandern können. Kleinere Proteine durchlaufen das Gel schneller als größere. Darüber hinaus profitiert die SDS-PAGE von den Prinzipien der Isotachophorese, wodurch Proteinproben zu engen, definierten Banden konzentriert werden und somit die Auflösung verbessert wird (in kontrollierten Laborbedingungen wird eine mindestens zehnfache Konzentration beobachtet).
Die sorgfältige Vorbereitung der Proben, die Zusammensetzung der Puffer und die Eigenschaften des Gels sind entscheidend für eine optimale Trennung und Reproduzierbarkeit. Die Bestandteile typischer Probenpuffer und ihre jeweiligen Funktionen sind in Tabelle 1 zusammengefasst.
Tabelle 1. Häufig verwendete Puffer zur Probenvorbereitung. Proteinort Puffer Zweck
| Protein-Lokalisierung | Puffer | Zweck der Komponenten (typische Konzentrationen in Klammern angegeben) |
| Ganze Zelle |
Nicht denaturierender, nichtionischer Puffer: NP-40-Puffer: 50 mM Tris pH 8,0, 150 mM NaCl, 1 % NP-40 oder TritonX-100 |
Tris-HCl (10–50 mM): Pufferung der Lösung (trägt zur Stabilität der Proteine bei) |
|
Denaturierungspuffer: RIPA-Puffer: 150 mM NaCl, 1 % NP-40 oder Triton X-100, 0,5 % Natrium Desoxycholat, 0,1 % SDS, 50 mM Tris, pH 8,0 |
Natriumdeoxycholat (0,1–0,5 %): ionisches Detergens, löst Zellen auf und macht Zell- und Membranbestandteile löslich SDS (0,1–1 %): ionisches Detergens, verursacht Membranzerstörung und linearisiert Proteine durch Bindung an diese |
|
| Zytoplasmatisch (löslich) |
20 mM Tris-HCl, pH 7,5 oder NP-40-Puffer | Siehe oben |
| Zytoplasmatisch (an das Zytoskelett gebunden) |
PIPES-Triton-Puffer: 10 mM PIPES, 50 mM KCl, 1 % Triton X-100, 10 mM EGTA, 3 mM MgCl2, 2 M Glycerin |
ROHRE (10 M): Pufferung der Lösung KCl (50–150 mM): reduziert unspezifische Bindungen, erhält die Ionenstärke EGTA (1–3 mM): Hemmung von Ca2+-Metalloproteasen MgCl2 (1–5 mM): reduziert unspezifische Bindungen, hat zusammen mit KCl eine stabilisierende Wirkung auf einige Komplexe Glycerin (1–10 %): stabilisiert Proteine |
| Membrangebundener | RIPA-Puffer | Siehe oben |
| Mitochondrien | RIPA-Puffer | Siehe oben |
| Nukleärer | RIPA-Puffer | Siehe oben; Hinweis: NP-40 und Triton X-100 können keine Kernmembranen lysieren |
| Proteinlokalisierung | Puffer |
Der erforderliche Gelanteil hängt von der Größe des Proteins ab, das Sie untersuchen möchten:
| Proteingröße | Gelanteil |
| 4–40 kDa | 20% |
| 12–45 kDa | 15% |
| 10–70 kDa | 12% |
| 15–100 kDa | 10% |
| 25–100 kDa | 8% |
| 60-210 kDa | 5% |
Gradientengele können ebenfalls verwendet werden.
| Proteingröße | Gelanteil |
| 5-200 kDa | 4-12 gradient |
| 4-200 kDa | 4-20 gradient |
| 3.5-110 kDa | 10-20 gradient |
Kontinuierliche versus diskontinuierliche Puffersysteme
Elektrophoretische Trennsysteme lassen sich grob in kontinuierliche und diskontinuierliche (auch multiphasische oder isotachophoretische genannt) Puffersysteme unterteilen, die sich jeweils durch unterschiedliche Pufferkonfigurationen und Trennungsdynamiken auszeichnen.
1. Kontinuierliche Puffersysteme:
-
Gleiche Pufferionen und pH-Wert in Probe, Gel und Reservoirs.
-
Gel aus einer einzigen Acrylamidkonzentration.
-
Proteine trennen sich direkt im Gel ohne Stapelung.
-
Wichtiger Punkt: Die Auflösung hängt stark von kleinen, konzentrierten Probenmengen ab.
2. Diskontinuierliche (mehrphasige) Puffersysteme:
-
Zwei verschiedene Puffer mit unterschiedlichem pH-Wert und unterschiedlicher Ionenstärke:
-
Stapelgel: pH-Wert ~6,8, niedriger Tris-HCl-Gehalt (0,125 M), große Poren.
-
Auflösungsgel: pH-Wert ~8,8, hoher Tris-HCl-Gehalt (0,375 M), kleine Poren.
-
-
Laufpuffer (z. B. Tris-Glycin) oberhalb des Stapelgels aufgetragen.
3. Trennmechanismus:
-
Cl⁻-Ionen wirken als Leitionen, Glycinat als Nachläuferionen.
-
Proteine konzentrieren sich zu dichten Banden im Stapelgel („Stapelungseffekt“).
-
Im Trenngel trennen sich die Proteine nach ihrem Molekulargewicht, da sie zu Hauptionen werden.
4. Vorteile diskontinuierlicher Systeme:
-
Ermöglicht das Laden größerer Mengen verdünnter Proben.
-
Überlegene Auflösung durch Stapelung und unterschiedliche Gelporengrößen.
-
Effektivere Trennung auf Basis des Molekulargewichts.
Proteintransfer
Nach der Elektrophorese werden die Proteine aus einem Polyacrylamidgel auf eine Membran übertragen. Dieser Schritt ist entscheidend, um die Proteine für die weitere Detektion und Analyse (z. B. Western Blotting) zu immobilisieren.
1. Membranen für den Proteintransfer
Es sind verschiedene Membranen mit jeweils spezifischen Vorteilen erhältlich:
-
Nitrocellulose ist die am häufigsten verwendete Membran. Sie hat eine hohe Proteinbindungsaffinität, kann sowohl Proteine als auch Glykoproteine immobilisieren und ist mit chemilumineszenten, chromogenen und fluoreszierenden Nachweismethoden kompatibel. Da sie jedoch aus mit Salpetersäure behandelter Cellulose gewonnen wird, ist sie spröde.
-
PVDF (Polyvinylidendifluorid) bietet eine bessere mechanische Festigkeit als Nitrocellulose und eignet sich daher ideal zum Abstreifen und erneuten Testen oder zum Sequenzieren von Proteinen. PVDF kann jedoch manchmal eine stärkere Hintergrundfärbung verursachen, sodass eine zusätzliche Optimierung erforderlich ist.
2. Methoden zum Proteintransfer
Proteine können mit verschiedenen Methoden vom Gel auf die Membran übertragen werden:
-
Kapillartransfer beruht auf passiver Kapillarwirkung.
-
Diffusionstransfer ermöglicht eine langsame Diffusion von Proteinen auf die Membran.
-
Elektroblotting ist die schnellste und vollständigste Transfermethode.
Unter diesen Methoden ist Elektroblotting aufgrund seiner Schnelligkeit und Effizienz die bevorzugte Methode.
Arten des Elektrobottings
Es gibt drei Arten von Elektroblotting, die sich nach der Art der Verwendung des Puffers unterscheiden:
-
Beim Trockenelektroblotting werden trockene Komponenten ohne Zugabe von Flüssigkeit verwendet.
- Beim halbtrockenen Elektrobotting werden mit Transferpuffer getränkte Filterpapiere zwischen die Membran und das Gel gelegt.
-
Beim Nass-Elektroblotting wird das gesamte Gel-Membran-Sandwich in einen Transferpuffer getaucht, was besonders für den Transfer großer Proteine empfohlen wird.
1. Grundlagen des Elektrobottings
Während des Elektrobottings:
-
Ein elektrisches Feld wird senkrecht zum Gel angelegt.
-
Proteine wandern aufgrund ihrer Ladung aus dem Gel auf die Membran.
-
Das Gel muss zur negativen Elektrode (Kathode) zeigen, während die Membran zur positiven Elektrode (Anode) zeigt.
-
Um eine ordnungsgemäße Übertragung zu gewährleisten, werden das Gel und die Membran fest aufeinander gelegt und mit Filterpapier und Fasermatten auf beiden Seiten abgedeckt, um Blasen zu vermeiden, die zu einer ungleichmäßigen Übertragung führen könnten.
a. Übertragungspuffer
Transferpuffer tragen dazu bei, die Proteinstruktur zu erhalten und die Bewegung während des Transfers zu verbessern. Zu den gängigen Puffern gehören:
-
Towbin-Puffer (25 mM Tris, 192 mM Glycin, 20 % Methanol, pH 8,3) wird häufig für den Nass-Transfer verwendet.
-
Für den halbtrockenen Transfer wird der Bjerrum-Schafer-Nielsen-Puffer (48 mM Tris, 39 mM Glycin, pH 9,2, 20 % Methanol) bevorzugt.
-
CAPS-Puffer (60 mM Tris und 40 mM CAPS) wird insbesondere für Proteine mit hohen isoelektrischen Punkten verwendet.
Methanol im Puffer verhindert das Aufquellen des Gels und verbessert die Proteinbindung an die Membran. In einigen Fällen können jedoch auch methanolfreie Puffer starke Detektionssignale erzeugen.
Die Rolle der Komponenten des Transferpuffers sind in Tabelle 2 beschrieben.
Tabelle 2. Zusammensetzung des Lauf- und Transferpuffers und Rolle der einzelnen Komponenten
| Potenzielle Komponente | Rolle |
| Laufende Puffer | |
| Acetate | Gute Trennung im höheren Massenbereich (100–500 kDa) |
| Glycine | Glycinat-Ionen bereitstellen, um während der Elektrophorese in einem Stacking-Gel ein hinteres Ende zu bilden. |
| MES | Erforderlich, um einen relativ konstanten pH-Wert aufrechtzuerhalten. Liefert MES-Ionen zur Bildung eines Nachlaufs während der Elektrophorese. MES führt zu einer größeren Trennung im unteren Massenbereich (<50 kDa). MES-SDS-Puffer ist aufgrund des im Vergleich zu MOPS niedrigeren pKa-Werts von MES schneller als MOPS-SDS-Puffer. |
| MOPS | Erforderlich, um einen relativ konstanten pH-Wert aufrechtzuerhalten. Liefert MOPS-Ionen zur Bildung eines Nachlaufs während der Elektrophorese. MOPS sorgt für eine bessere Trennung im mittleren bis höheren Massenbereich. Der Unterschied in der Bewegung der MES- und MOPS-Ionen beeinflusst die Stapelung, was zu einem Unterschied im Proteintrennungsbereich zwischen MES- und MOPS-Puffern führt. |
| SDS | Hilft dabei, Proteine in einer negativen Gesamtladung zu halten. |
| Tricine | Erforderlich, um einen relativ konstanten pH-Wert aufrechtzuerhalten. Tricin ersetzt Glycin im Laufpuffer und liefert Tricin-Ionen, die während der Elektrophorese eine Endgruppe bilden. Tricin sorgt für eine effizientere Stapelung und Auflösung von Proteinen mit niedrigem Molekulargewicht. Gut geeignet für die Trennung von Proteinen mit niedrigem Molekulargewicht (0,5–50 kDa). |
| Tris-HCl | Erforderlich, um einen relativ konstanten pH-Wert aufrechtzuerhalten. Liefert Chloridionen, die während der Elektrophorese im Stacking-Gel die führende Endgruppe bilden. Wasserstoffionen leiten Elektrizität. |
| Transfer buffers | |
| Tris-HCl | Erforderlich, um einen relativ konstanten pH-Wert aufrechtzuerhalten |
| Glycine | In Abwesenheit von Methanol verhindert es das Aufquellen des Gels. |
| Methanol | Verhindert das Aufquellen des Gels während des Transfers und verbessert die Proteinbindung an Nitrocellulose. Entfernt SDS aus Proteinen und ermöglicht so eine bessere Bindung an Nitrocellulosemembranen. Einige Labore verzichten mittlerweile auf Methanol in Transferpuffern, da die Ergebnisse ohne Methanol nicht signifikant von denen mit Methanol abweichen. In einigen Protokollen wird 10 % Ethanol anstelle von 20 % Methanol verwendet. |
| SDS | SDS (bis zu 0,1 %) im Transferpuffer erhöht die Proteintransfereffizienz, insbesondere bei großen Proteinen, kann jedoch die Menge der an die Membran gebundenen Proteine verringern. Bei Nitrocellulosemembranen mit größeren Porengrößen (0,45 μm) können kleine SDS-denaturierte Proteine die Membran passieren. |
| CAPS | Erforderlich, um einen relativ konstanten pH-Wert aufrechtzuerhalten. Empfohlen für den Transfer von Proteinen mit hohem Molekulargewicht (>150 kDa) |
| CAPS: (Cyclohexylamino)-L-propansulfonsäure; MES: 2-(N-Morpholino)ethansulfonsäure; MOPS: 3-(N-Morpholino)propansulfonsäure; SDS: Natriumdodecylsulfat; Tris-HCl: Tris-Hydrochlorid. |
|
b. Wichtige Überlegungen
-
Proteine mit sehr geringem Molekulargewicht (<10 kDa) können möglicherweise nur unzureichend übertragen werden.
-
Wenn der pH-Wert des Transferpuffers niedriger ist als der isoelektrische Punkt des Proteins, können Proteine rückwärts wandern.
-
Für die effiziente Übertragung von Proteinen mit einem hohen isoelektrischen Punkt werden häufig spezielle Puffer (wie CAPS-basierte Puffer) benötigt.
Blockierungsschritt beim Western Blotting
Nach dem erfolgreichen Proteintransfer auf die Membran ist das Blockieren ein wichtiger Schritt, um die Spezifität und Klarheit der anschließenden Immunodetektion sicherzustellen.

1. Zweck und Bedeutung
-
Die Blockierung verhindert die unspezifische Bindung von Antikörpern an die Membran, wodurch Hintergrundrauschen minimiert und falsch-positive Signale eliminiert werden.
-
Es verbessert die Genauigkeit und Zuverlässigkeit der Protein-Detektion beim Western Blotting.
2. Sperrverfahren
-
Die Membran wird 1 Stunde lang bei Raumtemperatur in einer Blockierungslösung inkubiert, die Folgendes enthält:
-
Rinderserumalbumin (BSA) oder fettfreie Trockenmilch, verdünnt in:
-
Tris-gepufferte Kochsalzlösung mit 0,1 % Tween 20 (TBST) oder
-
Phosphatgepufferte Kochsalzlösung mit 0,1 % Tween 20 (PBST).
-
-
- Die in BSA und Milch enthaltenen Proteine (vor allem Kasein und Molkenproteine) binden sich an freie Stellen der Membran und verhindern so unspezifische Wechselwirkungen zwischen der Membran und den primären oder sekundären Antikörpern.
3. Membraneigenschaften und Rolle von Tween 20
- Nitrocellulose und PVDF-Membranen weisen vorwiegend durch hydrophobe Wechselwirkungen eine hohe Proteinbindungsaffinität auf.
-
Während BSA und Milchproteine eine schwache Bindung mit den übertragenen Proteinen eingehen können, reduziert die Zugabe von Tween 20 im Blocking-Puffer diese unspezifischen Wechselwirkungen und gewährleistet so eine effektivere Blockierung.
Antikörperinkubation und -nachweis im Western Blotting
1. Die Rolle der Antikörperinkubation beim Western Blotting
Nach dem Blockieren der Membran, um eine unspezifische Bindung zu verhindern, geht der Western Blot zum entscheidenden Schritt der Antikörperinkubation über. Zunächst wird die Membran mit einem Primärantikörper inkubiert, der spezifisch an das Zielprotein bindet. Nach dem Abwaschen der ungebundenen Antikörper mit Puffern wie TBST oder PBST wird ein Sekundärantikörper zugegeben. Dieser Sekundärantikörper erkennt den Primärantikörper und ist in der Regel mit einem Enzym wie HRP (Horseradish Peroxidase) oder Alkalische Phosphatase (AP) konjugiert, wodurch eine anschließende Signalerkennung ermöglicht wird.
-
Primäre Antikörper: Unverzichtbar für die spezifische und selektive Bindung an Zielproteine.
- Sekundäre Antikörper: Ermöglichen den Nachweis und die Verstärkung des Signals.
Tipp: Es ist wichtig, die Antikörperkonzentration zu optimieren, um die Empfindlichkeit zu maximieren und den Hintergrund zu reduzieren. Unser technischer Support hilft Ihnen gerne bei der Protokolloptimierung.
Waschschritte sind entscheidend, um ungebundene Antikörper zu entfernen. Übermäßiges Waschen (>20 Minuten) kann jedoch das Signal schwächen – daher ist ein ausgewogenes Timing entscheidend!
2. Signalerkennungsmethoden
Sobald das Zielprotein durch markierte Antikörper gebunden ist, muss das Signal nachgewiesen werden. Dazu stehen mehrere Methoden zur Verfügung:
a. Chemilumineszenz (ECL)
Die verstärkte Chemilumineszenz (ECL) ist die beliebteste Nachweismethode. Unter Verwendung von HRP-konjugierten Sekundärantikörpern und ECL-Substraten wird Licht erzeugt und entweder auf Film oder mit einem digitalen Bildgeber erfasst.
-
Hochsensibel
-
Großer Dynamikbereich
-
Ideal für den Nachweis von Proteinen mit geringer Häufigkeit
Empfohlene Produkte:
HRP-konjugierte Sekundärantikörper
b. Fluoreszenzdetektion
Fluoreszenzfarbstoff-konjugierte Sekundärantikörper ermöglichen eine multiplexe Detektion – das heißt, Sie können mehrere Proteine gleichzeitig mit verschiedenen Fluorophoren sichtbar machen.
- Geeignet für die gleichzeitige Erkennung mehrerer Ziele
-
Hervorragender Dynamikbereich und Quantifizierung
-
Am besten bei Verwendung von Fluorophoren im Infrarotbereich
Empfohlene Produkte:
Sekundäre Antikörper (sichtbarer und infraroter Bereich)
c. Chromogene Detektion
Chromogene Methoden nutzen Enzymreaktionen, um einen farbigen Niederschlag auf der Membran zu erzeugen, der mit bloßem Auge sichtbar ist.
- Ideal, wenn die Bildgebungsausrüstung begrenzt ist.
-
Einfache Visualisierung
-
Keine spezielle Ausrüstung erforderlich
-
Ideal für Ausbildungslabore und Grundlagenforschung
Empfohlene Produkte:
Chromogene Substrat-Kits (e.g., BCIP/NBT)
d. Radioaktive Detektion (heute weniger verbreitet)
Radioaktive Sonden wurden früher häufig für Western Blots verwendet, sind heute jedoch weitgehend durch sicherere, ebenso empfindliche Methoden wie ECL und Fluoreszenz ersetzt worden. Die Auswahl der geeigneten Nachweismethode ist entscheidend für den Erfolg Ihres Western Blots und hängt von der Empfindlichkeit, Spezifität und Art der Analyse ab, die Sie benötigen:
-
Maximale Empfindlichkeit? → Wählen Sie die erweiterte Chemilumineszenz (ECL)-Detektion.
ECL bietet eine hohe Empfindlichkeit und einen breiten Dynamikbereich und eignet sich daher ideal für den Nachweis von Proteinen in geringen Konzentrationen. Die Signalintensität kann auf Röntgenfilmen oder digitalen Bildgebungssystemen erfasst werden. -
Multiplex-Analyse (gleichzeitiger Nachweis mehrerer Ziele)? → Wählen Sie fluoreszenzbasierte Detektion.
Fluoreszierende Sekundärantikörper ermöglichen den gleichzeitigen Nachweis mehrerer Proteine, ohne dass die Membranen entfernt und erneut untersucht werden müssen. Die Signale sind äußerst stabil, quantitativ und ideal für vergleichende Studien. -
Einfache, schnelle Visualisierung? → Wählen Sie chromogene Detektion..
- Chromogene Systeme nutzen Enzym-Substrat-Reaktionen, um direkt auf der Membran einen farbigen Niederschlag zu erzeugen. Sie sind zwar weniger empfindlich als ECL oder Fluoreszenz, dafür aber einfach zu verwenden und erfordern keine spezielle Bildgebungsausrüstung.
Darüber hinaus müssen die Auswahl der Primär- und Sekundärantikörper sowie der Substrate oder Nachweisreagenzien mit der von Ihnen gewählten Nachweismethode kompatibel sein. Die richtige Kombination ist entscheidend, um spezifische und starke Signale zu erzielen und gleichzeitig Hintergrundrauschen zu minimieren..