
Protokoll für die Polymerase-Kettenreaktion (PCR)
Einführung
Die Polymerase-Kettenreaktion (PCR) ist eine grundlegende Technik in der Molekularbiologie, die die Amplifikation spezifischer DNA-Sequenzen aus geringen Mengen an Ausgangsmaterial ermöglicht. Seit ihrer Erfindung durch Kary Mullis im Jahr 1983 hat die PCR die genetische Forschung, Diagnostik, Forensik und Biotechnologie revolutioniert. Dieses Protokoll bietet eine umfassende Anleitung zur Durchführung einer Standard-PCR, einschließlich detaillierter Schritte, Reagenzspezifikationen, Fehlersuche und Optimierungsstrategien.
Prinzip der PCR
Die PCR ist ein enzymatischer Prozess, der ein spezifisches DNA-Segment durch wiederholte Zyklen von Denaturierung, Annealing und Extension amplifiziert. Die Reaktion basiert auf einer thermostabilen DNA-Polymerase, typischerweise Taq-Polymerase, die hohen Temperaturen standhält, die zur Denaturierung doppelsträngiger DNA erforderlich sind (Abbildung 1).
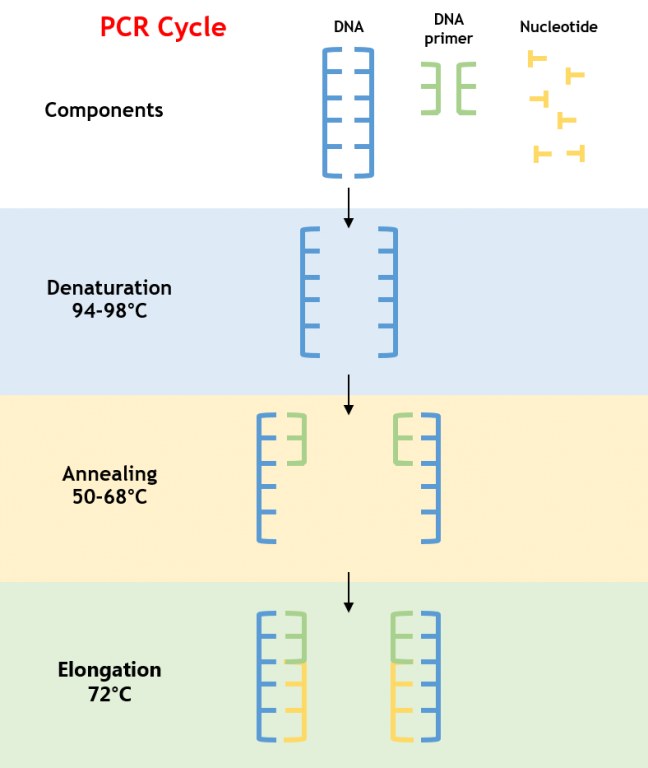
Abbildung 1: PCR-Amplifikationsprozess
- Denaturierung: Die DNA wird auf 94–98°C erhitzt, wodurch Wasserstoffbindungen aufgebrochen werden und einzelsträngige DNA entsteht.
- Annealing: Die Temperatur wird auf 50–68°C gesenkt, sodass Primer an komplementäre Sequenzen, die das Ziel-DNA flankieren, binden können.
- Extension: Die Temperatur wird auf 72°C erhöht, was für die Taq-Polymerase optimal ist, um neue DNA-Stränge durch das Hinzufügen von Deoxynukleotidtriphosphaten (dNTPs) zu synthetisieren.
Materialien und Ausrüstung
Stellen Sie vor Beginn sicher, dass alle Reagenzien und Geräte verfügbar sind und ordnungsgemäß gelagert werden. Verwenden Sie Reagenzien in Molekularbiologie-Qualität, um Kontaminationen oder Hemmungen zu vermeiden.
Reagenzien
- Taq-DNA-Polymerase: Bei -20°C lagern.
- 10X PCR-Puffer: Enthält typischerweise 100 mM Tris-HCl, 500 mM KCl und 15 mM MgCl₂ (pH 8,3–8,8). Einige Puffer enthalten kein MgCl₂, was eine separate Zugabe erfordert. Bei -20°C oder 4°C gemäß Herstellerangaben lagern.
- Magnesiumchlorid: 25 mM MgCl₂-Lösung (falls nicht im PCR-Puffer enthalten). Bei -20°C oder 4°C lagern.
- dNTPs: Deoxynukleotidtriphosphate (dATP, dTTP, dCTP, dGTP; je 10 mM, kombiniert oder getrennt). Bei -20°C lagern.
- Primer: Vorwärts- und Rückwärtsprimer (100 µM Stammlösung in TE-Puffer, verdünnt auf 10 µM Arbeitslösung).
- Template-DNA: Genomische DNA (10–100 ng), Plasmid-DNA (0,1–1 ng) oder cDNA (1–10 ng), je nach Anwendung. Bei -20°C lagern.
- Nukleasefreies Wasser: Steriles Wasser in Molekularbiologie-Qualität. Bei Raumtemperatur lagern.
- Master-Mix
- SYBR® Safe DNA-Gelfarbstoff: Für die Visualisierung von Agarosegelen (Alternative: Ethidiumbromid, aber aufgrund der Toxizität mit Vorsicht verwenden). Bei 4°C lagern.
- 6X-Ladefarbstoff: Für die Gelelektrophorese (z. B. enthält Bromphenolblau und Xylencyanol). Bei 4°C lagern.
- DNA-Leiter: 100 bp oder 1 kb Leiter, abhängig von der erwarteten Amplikongröße. Bei -20°C lagern.
Ausrüstung
- Thermocycler: Programmierbares PCR-Gerät.
- PCR-Röhrchen: 0,2 mL dünnwandige PCR-Röhrchen oder 96-Well-Platten, kompatibel mit dem Thermocycler.
- Pipetten und Spitzen: Kalibrierte Pipetten (P2, P10, P20, P200, P1000) mit Filterspitzen, um Kontaminationen zu vermeiden.
- Mikrozentrifuge: Für kurze Zentrifugationen, um Reaktionskomponenten zu sammeln.
- Agarose-Gelelektrophorese-System: Einschließlich Gussform, Kamm, Stromversorgung und UV-Transilluminator (oder Blaulicht für SYBR Safe).
- Vortex-Mixer: Zum Mischen von Reagenzien.
- Eiskübel: Um Reagenzien während des Aufbaus kühl zu halten.
- Spektrophotometer: Zur Quantifizierung der DNA-Konzentration.
PCR-Protokoll
Schritt 1: Primer-Design und Vorbereitung
Primer sind kurze, einzelstrangige DNA-Oligonukleotide (18–25 Basen), die die Zielsequenz flankieren und als Ausgangspunkte für die DNA-Synthese dienen. Ein korrektes Primer-Design ist entscheidend für eine spezifische und effiziente Amplifikation.
Richtlinien für das Primer-Design
Verwenden Sie Softwaretools zum Design von Primern. Beachten Sie folgende Kriterien:
- Länge: 18–25 Basen. Kürzere Primer (<18) können an Spezifität verlieren; längere Primer (>25) können Sekundärstrukturen bilden.
- Schmelztemperatur (Tm): 55–65°C, wobei Vorwärts- und Rückwärtsprimer innerhalb von 2°C liegen sollten. Berechnen Sie Tm mit der Formel für eine grobe Schätzung oder verwenden Sie Software für Genauigkeit (berücksichtigt Salz- und Primerkonzentration).
Tm = 4 (G+C) + 2 (A+T)
- GC-Gehalt: 40–60%. Vermeiden Sie Extreme (<30% oder >70%), um schlechtes Annealing oder Sekundärstrukturen zu verhindern.
- 3'-Ende: Enden Sie mit einem G oder C (stärkere Basenpaarung) und vermeiden Sie Sequenzen von drei oder mehr identischen Basen (z. B. GGG), um Fehlpriming zu reduzieren.
- Spezifität: Überprüfen Sie die Spezifität mit NCBI BLAST (blast.ncbi.nlm.nih.gov), um sicherzustellen, dass Primer nur an die Zielsequenz binden. Vermeiden Sie Homologie zu nicht-zielgerichteten Regionen.
- Sekundärstrukturen: Vermeiden Sie Haarnadeln, Selbst-Dimere und Hetero-Dimere. Delta G für Dimere sollte >-5 kcal/mol betragen.
- Amplikongröße: Entwerfen Sie für 100–1000 bp Amplikons für Standard-PCR. Längere Amplikons (bis zu 5 kb) sind möglich, erfordern jedoch Optimierung.
- Vermeidung von Wiederholungen: Platzieren Sie keine Primer in Regionen mit repetitiven Sequenzen oder niedriger Komplexität (z. B. ATATAT).
Primer-Synthese und Lagerung
- Bestellung: Bestellen Sie HPLC-gereinigte Primer. HPLC-Reinigung gewährleistet hohe Reinheit und reduziert unspezifische Amplifikation.
- Resuspension: Resuspendieren Sie lyophilierte Primer in TE-Puffer (10 mM Tris-HCl, 1 mM EDTA, pH 8,0) zu einer 100 µM Stammlösung.
- Arbeitslösung: Verdünnen Sie die Stammlösung auf 10 µM in nukleasefreiem Wasser. Bei -20°C lagern.
- Lagerung: Lagern Sie 100 µM Stammlösungen bei -20°C für bis zu 1 Jahr; 10 µM Arbeitslösungen sind bei -20°C für 6 Monate oder bei 4°C für 1 Monat stabil. Vermeiden Sie wiederholte Gefrier-Tau-Zyklen (>10), um Abbau zu verhindern.
Primer-Qualitätskontrolle
- Quantifizierung: Überprüfen Sie die Primer-Konzentration mit einem Spektrophotometer (A260). Erwartete Absorption für 100 µM liegt bei ~0,3–0,5, abhängig von der Sequenz.
- Integrität: Optional – führen Sie Primer auf einem 2% Agarosegel durch, um sicherzustellen, dass kein Abbau vorliegt (sollte als einzelne, scharfe Bande erscheinen).
- Test-Amplifikation: Führen Sie eine Test-PCR mit einer bekannten Template-Primer-Kombination durch, um die Funktionalität der Primer vor groß angelegten Experimenten zu bestätigen.
Schritt 2: Reaktionsaufbau
Die PCR-Reaktionsmischung enthält alle Komponenten, die für die DNA-Amplifikation erforderlich sind. Das angegebene Protokoll ist auf ein Reaktionsvolumen von 50 µL ausgelegt, mit einem Master-Mix für 11 Reaktionen (10 Proben + 1 Kontrolle ohne Template) zur Berücksichtigung von Pipettierverlusten.
Reaktionskomponenten
Die folgende Tabelle listet die Reagenzien und Volumina für eine 50 µL-Reaktion und den Master-Mix für 11 Reaktionen auf.
| Reagenz | Volumen pro Reaktion (µL) | Gesamt für 11 Reaktionen (µL) |
| Nukleasefreies Wasser | 36,5 | 401,5 |
| 10X PCR-Puffer | 5,0 | 55,0 |
| dNTPs (je 10 mM) | 1,0 | 11,0 |
| MgCl₂ (25 mM) | 3,0 | 33,0 |
| Vorwärtsprimer (10 µM) | 1,0 | 11,0 |
| Rückwärtsprimer (10 µM) | 1,0 | 11,0 |
| Taq-Polymerase (5 U/µL) | 0,5 | 5,5 |
| Template-DNA (variabel) | 3,0 | Separat hinzufügen |
Schritt 3: Thermisches Zyklieren
Der Thermocycler unterzieht die Reaktion wiederholten Temperaturwechseln, um Denaturierung, Annealing und Extension zu ermöglichen. Die angegebenen Zyklusbedingungen sind für Standard-PCR mit Taq-Polymerase und Amplikons von 100–1000 bp optimiert.
|
|
|
|
|
NeoCycler Trio - Multi-Block-Thermocycler (Kat.-Nr. NB-12-3024) NeoCycler Duo - Multi-Block-Thermocycler (Kat.-Nr. NB-12-3025)
|
NeoCycler 300 - Schneller Gradienten-Thermocycler (mit 96/77 / 96 Modul) (Kat.-Nr. NB-12-3001) NeoCycler 200 - Gradienten-Thermocycler (Kat.-Nr. NB-12-3002) NeoCycler 100 - Nicht-Gradienten-Thermocycler (mit 96/77 / 96 Modul) (Kat.-Nr. NB-12-3003) |
|
|
|
|
|
|
NeoCycler 600 - Super-Gradienten-Thermocycler (Kat.-Nr. NB-12-3026) |
NeoCycler Mini (Kat.-Nr. NB-12-3029-2) |




Thermisches Zyklusprogramm
Programmieren Sie den Thermocycler mit den folgenden Schritten:
- Initiale Denaturierung: 95°C für 2 Minuten
- Denaturiert doppelstrangige Template-DNA und aktiviert Taq-Polymerase (Hot-Start-Versionen können länger benötigen, z. B. 5–10 Minuten).
- Zyklus (30–35 Zyklen):
- Denaturierung: 95°C für 30 Sekunden
- Trennt DNA-Stränge. Kürzere Zeiten (15–20 Sekunden) können für kleine Templates (z. B. Plasmide) ausreichen.
- Annealing: 55–65°C für 30 Sekunden
- Primer binden an komplementäre Sequenzen. Stellen Sie 2–5°C unter der niedrigeren Primer-Tm ein (z. B. bei Tm von 60°C, verwenden Sie 55–58°C). Optimieren Sie bei Bedarf (siehe Abschnitt 8).
- Extension: 72°C für 1 Minute pro kb
- Taq-Polymerase synthetisiert neue DNA. Für ein 500 bp Amplikons verwenden Sie 30–60 Sekunden; für 1 kb verwenden Sie 60 Sekunden. Minimum 30 Sekunden für <500 bp.
- Denaturierung: 95°C für 30 Sekunden
- Finale Extension: 72°C für 5 Minuten
- Vervollständigt partielle Extensionen und fügt 3'-A-Überhänge hinzu (nützlich für TA-Klonierung).
- Halt: 4°C unbegrenzt
- Speichert Reaktionen bis zur Entnahme. Vermeiden Sie längere Lagerung (>24 Stunden), um Abbau zu verhindern.
Zykluszahl
- Standard: 30 Zyklen für moderate Template-Mengen (10–50 ng genomische DNA).
- Niedrige Template-Menge: 35 Zyklen für geringe Template-Mengen (<1 ng) oder cDNA.
- Hohe Template-Menge: 25–28 Zyklen für hohe Template-Mengen (>100 ng), um unspezifische Produkte zu reduzieren.
- Übermäßige Zyklen: >35 Zyklen können unspezifische Amplifikation oder Primer-Dimere verstärken.
Einstellungen des Thermocyclers
- Deckel-Temperatur: Auf 105°C einstellen, um Kondensation zu verhindern.
- Rampenrate: Verwenden Sie Standardeinstellungen (2–5°C/Sekunde). Schnellere Rampen können bei älteren Geräten Anpassungen erfordern.
- Volumen: Stellen Sie im Cycler-Programm 50 µL ein, um eine genaue Erwärmung sicherzustellen.
Variationen
- Kurze Amplikons (<200 bp): Reduzieren Sie die Extensionszeit auf 15–30 Sekunden.
- Lange Amplikons (1–5 kb): Erhöhen Sie die Extensionszeit (1–2 Minuten/kb) und erwägen Sie hochpräzise Polymerasen.
- GC-reiche Templates: Verwenden Sie höhere Denaturierungstemperaturen (98°C) oder Additive wie DMSO (5–10%).
Schritt 4: Analyse der PCR-Produkte
Nach der PCR analysieren Sie die Produkte, um den Erfolg der Amplifikation und die Amplikongröße zu bestätigen. Die Agarose-Gelelektrophorese ist die Standardmethode.
Vorbereitung des Agarosegels
- Gelkonzentration:
- 1% Agarose für 500–5000 bp Amplikons.
- 1,5–2% Agarose für 100–500 bp Amplikons.
- Gel vorbereiten:
- Wiegen Sie Agarose ab.
- Lösen Sie sie in 1X TAE-Puffer (40 mM Tris, 20 mM Essigsäure, 1 mM EDTA) durch Erhitzen in der Mikrowelle, bis die Lösung klar ist.
- Kühlen Sie auf ~50°C ab, fügen Sie SYBR Safe (1 µL pro 10 mL Gel, gemäß Hersteller) hinzu und gießen Sie es in eine Gussform mit Kamm.
- Lassen Sie es 20–30 Minuten aushärten.
- Aufbau: Platzieren Sie das Gel in einem Elektrophoresetank, gefüllt mit 1X TAE-Puffer.
Probenvorbereitung
- Probe mischen: Kombinieren Sie 10 µL PCR-Produkt mit 2 µL 6X Ladefarbstoff (endgültig 1X).
- Leiter laden: Laden Sie 5–10 µL DNA-Leiter (100 bp oder 1 kb, abhängig von der Amplikongröße) in eine Spur.
- Proben laden: Laden Sie 12 µL jeder PCR-Produkt + Farbstoff-Mischung in separate Wells. Schließen Sie die Kontrolle ohne Template (NTC) ein, um Kontaminationen zu überprüfen.
Elektrophorese
- Gel laufen lassen: Lassen Sie das Gel bei 80–120 V für 30–60 Minuten laufen, bis der Ladefarbstoff (Bromphenolblau) etwa 2/3 der Gellänge gewandert ist.
- Visualisieren: Verwenden Sie einen UV-Transilluminator (oder Blaulicht für SYBR Safe), um Banden sichtbar zu machen. Fotografieren Sie das Gel zur Dokumentation.
Interpretation
- Erwartete Bande: Vergleichen Sie die Bandengröße mit der DNA-Leiter. Sie sollte der vorhergesagten Amplikongröße (basierend auf dem Primer-Design) entsprechen.
- NTC: In der NTC-Spur sollten keine Banden erscheinen. Banden deuten auf Kontamination oder Primer-Dimere hin.
- Schmieren: Weist auf unspezifische Amplifikation, degradierte Template oder überschüssige DNA hin.
- Keine Banden: Deutet auf fehlgeschlagene Amplifikation hin.
Nachanalyse
- Lagerung: Lagern Sie PCR-Produkte bei -20°C für Wochen oder bei 4°C für 1–2 Tage.
- Reinigung: Für nachgelagerte Anwendungen (z. B. Sequenzierung, Klonierung) reinigen Sie die Produkte mit einem PCR-Aufreinigungskit oder Gel-Extraktionskit.
- Quantifizierung: Messen Sie die Produktkonzentration bei Bedarf mit einem Nanodrop oder Fluorometer.
Qualitätskontrolle und Validierung
Um zuverlässige Ergebnisse zu gewährleisten, führen Sie an jedem Schritt eine Qualitätskontrolle durch.
Template-Qualität
- Reinheit: Ein A260/A280-Verhältnis von 1,8–2,0 zeigt reine DNA an. Niedrigere Verhältnisse deuten auf Protein- oder RNA-Kontamination hin.
- Integrität: Führen Sie Template-DNA auf einem 0,8% Agarosegel durch, um sicherzustellen, dass kein Abbau vorliegt (genomische DNA sollte als hochmolekulare Bande erscheinen).
- Konzentration: Verwenden Sie 10–100 ng genomische DNA oder 0,1–1 ng Plasmid-DNA pro 50 µL Reaktion.
Reagenzien-Qualität
- dNTPs: Überprüfen Sie auf Abbau (bei -20°C lagern, vermeiden Sie >10 Gefrier-Tau-Zyklen).
- Taq-Polymerase: Überprüfen Sie die Aktivität durch Testen mit einem bekannten Template-Primer-Paar.
- Primer: Bestätigen Sie Konzentration und Integrität.
Kontrollen
- NTC: Erkennt Kontaminationen oder Primer-Dimere.
- Positive Kontrolle: Verwenden Sie ein bekanntes Template-Primer-Paar, um die Reaktionsbedingungen zu bestätigen.
- Negative Kontrolle (optional): Lassen Sie Polymerase weg, um unspezifische Amplifikation zu überprüfen.
Gel-Analyse
- Leiter: Stellen Sie sicher, dass die Leiter klar auflöst, um die Gelqualität zu bestätigen.
- Bandenintensität: Starke, einzelne Banden zeigen eine erfolgreiche Amplifikation an. Schwache oder mehrere Banden deuten auf Optimierungsbedarf hin.
Fehlersuche
Die PCR kann Optimierung erfordern, um spezifische, hochproduktive Amplifikation zu erreichen. Nachfolgend sind häufige Probleme und Lösungen aufgeführt.
Keine Amplifikation
- Ursache: Niedrige Template-Konzentration, degradierte Template, falsche Primer oder suboptimale Annealing-Temperatur.
- Lösungen:
- Erhöhen Sie die Template-Menge (bis zu 200 ng) oder Zyklen (bis zu 35).
- Überprüfen Sie die Template-Integrität auf einem Gel.
- Bestätigen Sie Primer-Sequenzen und Tm; bei Bedarf neu gestalten.
- Führen Sie eine Gradienten-PCR (50–65°C Annealing) durch, um die optimale Temperatur zu finden.
- Überprüfen Sie die Taq-Polymerase-Aktivität mit einer positiven Kontrolle.
Unspezifische Banden
- Ursache: Niedrige Annealing-Temperatur, hohe MgCl₂-Konzentration, überschüssige Primer oder unspezifische Primer-Bindung.
- Lösungen:
- Erhöhen Sie die Annealing-Temperatur in 1–2°C-Schritten.
- Reduzieren Sie MgCl₂ auf 1–1,2 mM.
- Verringern Sie die Primer-Konzentration auf 0,1–0,15 µM.
- Gestalten Sie Primer für höhere Spezifität neu (verwenden Sie BLAST).
- Reduzieren Sie die Zykluszahl auf 25–28.
Schmieren
- Ursache: Überschüssige Template, degradierte Template oder Überzyklisierung.
- Lösungen:
- Reduzieren Sie die Template-Menge auf 10–50 ng.
- Überprüfen Sie die Template-Integrität.
- Verringern Sie die Zyklen auf 25–30.
- Verwenden Sie frische dNTPs und Polymerase.
Primer-Dimere
- Ursache: Primer, die Selbst- oder Hetero-Dimere bilden, sichtbar als niedermolekulare Banden (<100 bp) in der NTC.
- Lösungen:
- Gestalten Sie Primer neu, um Dimerbildung zu minimieren.
- Reduzieren Sie die Primer-Konzentration auf 0,1 µM.
- Erhöhen Sie die Annealing-Temperatur.
- Verwenden Sie Hot-Start Taq-Polymerase, um unspezifisches Priming während des Aufbaus zu verhindern.
Optimierungsstrategien
- Gradienten-PCR: Testen Sie einen Bereich von Annealing-Temperaturen (z. B. 50–65°C) in einem Durchgang, wenn der Cycler eine Gradientenfunktion unterstützt.
- MgCl₂-Titration: Testen Sie 1–4 mM MgCl₂ in 0,5 mM-Schritten.
- Additive: Für GC-reiche Templates fügen Sie 5–10% DMSO oder Betain (1–2 M) zur Reaktion hinzu.
- Hot-Start-PCR: Verwenden Sie Hot-Start Taq (z. B. Platinum Taq), um unspezifische Amplifikation während des Aufbaus zu reduzieren.
- Touchdown-PCR: Beginnen Sie das Annealing bei 5–10°C über Tm, senken Sie es um 0,5°C pro Zyklus für 10 Zyklen, und fahren Sie dann bei der niedrigeren Temperatur fort. Erhöht die Spezifität.
Fazit
Dieses umfassende PCR-Protokoll bietet einen robusten Rahmen für die Amplifikation von DNA mit hoher Spezifität und Ausbeute. Durch das Befolgen der detaillierten Schritte für Primer-Design, Reaktionsaufbau, thermisches Zyklieren und Produktanalyse können Benutzer zuverlässige Ergebnisse für eine Vielzahl von Anwendungen erzielen. Optimierungs- und Fehlersuchtipps gewährleisten Erfolg auch bei anspruchsvollen Templates oder Primern. Für fortgeschrittene Anwendungen passen Sie das Protokoll wie beschrieben an und konsultieren Sie die Herstellerangaben für spezialisierte Reagenzien oder Ausrüstung.